
研究背景
反应物在固体表面的吸附行为是研究反应机理的关键。在化学链燃烧中,铁基载氧体通过氧化还原反应传递晶格氧实现燃料高效利用。过渡金属掺杂可显著提升其反应活性,但掺杂引起的微观结构变化对H₂、CH₄、CO 等小分子燃料吸附行为的影响机制尚不明确。本研究采用 DFT 计算,系统研究气体分子在纯相及过渡金属掺杂铁基载氧体表面的吸附能、电子结构和相互作用机制。
研究内容
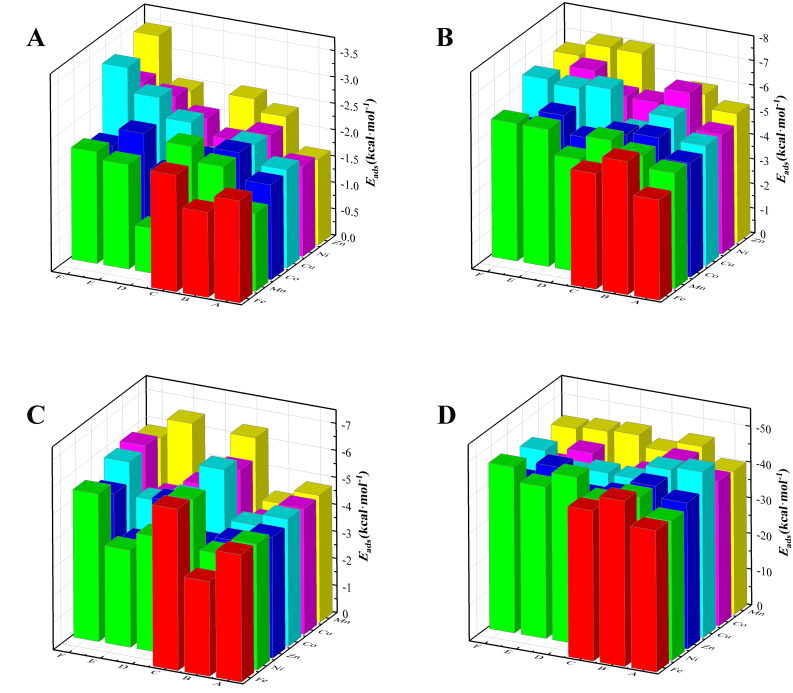
采用 AMS 计算软件中 BAND 模块中完成 H2、CO、CH4 等分子在过渡金属掺杂的 α-Fe2O3 模型表面的吸附行为计算。获得了反应物在载氧体表面最稳定的吸附构型。EDA 揭示了 H2、CH4 和 CO 在纯相及过渡金属掺杂载氧体表面的相互作用能范围分别为:H2 为 -1.33 至 -3.48 kcal/mol,CH4 为 -4.59 至 -7.26 kcal/mol,CO 物理吸附为 -3.37 至 -6.19 kcal/mol,化学吸附为 -37.68 至 -49.45 kcal/mol。其中,H2 与CO 的最优吸附位点为桥位,吸附能分别为 -2.214 与 -5.973 kcal/mol。CH4 的最稳定吸附为空心位,吸附能为 -5.420 kcal/mol。吸附能顺序为:化学吸附 CO > 物理吸附 CO > CH4 > H2。
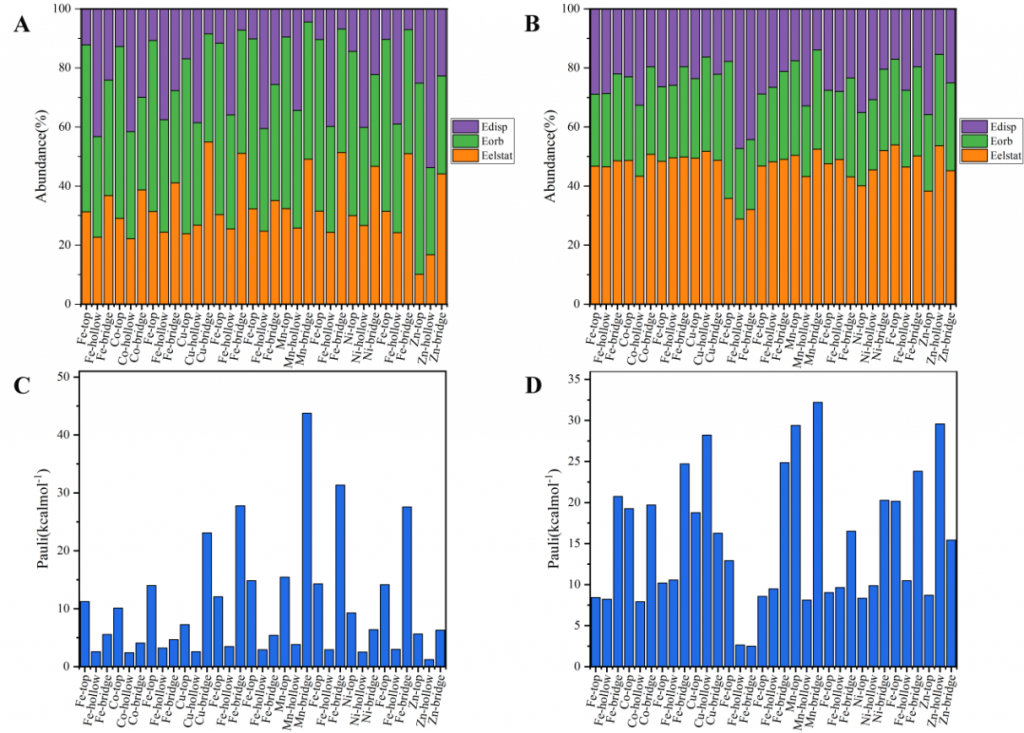
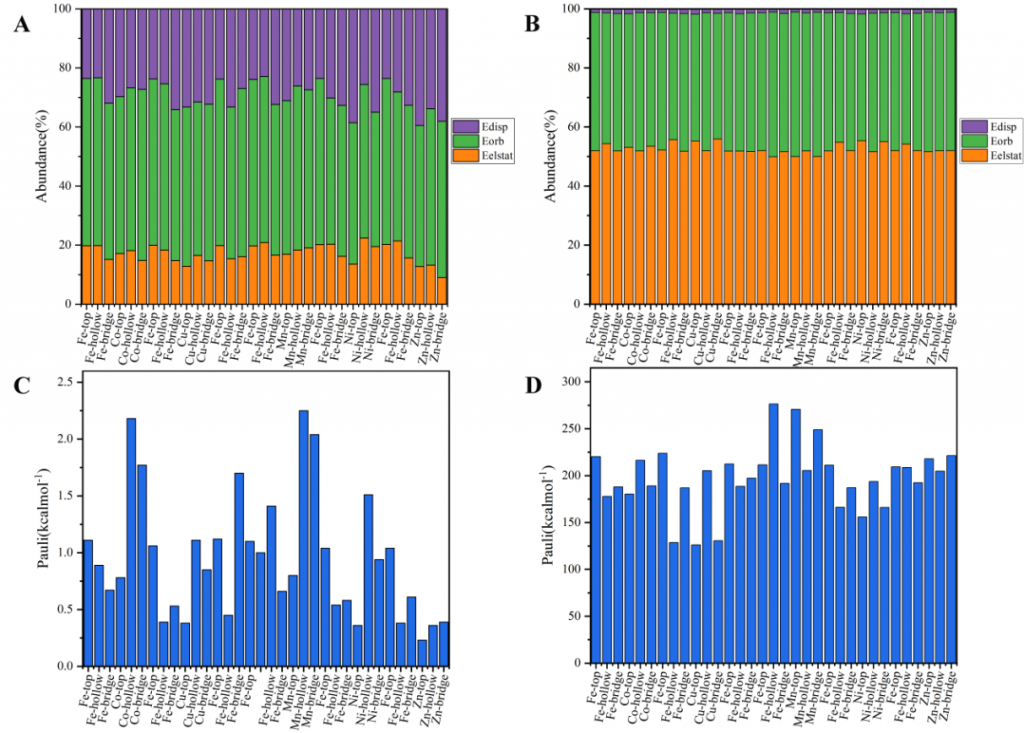
采用能量分解分析(EDA)方法将总相互作用能解构为不同能量分量的贡献,定量阐明了三种分子吸附的物理本质与主导作用力。CO 化学吸附的泡利排斥能(100-200 kcal/mol)显著强于物理吸附(1-2 kcal/mol),这归因于增强的排斥相互作用和空间效应。H2 吸附以轨道相互作用为主(55~65%)。CH₄ 体系中静电相互作用最关键(40~50%)。CO 物理吸附以轨道作用主导,化学吸附则同时依赖轨道与静电作用。
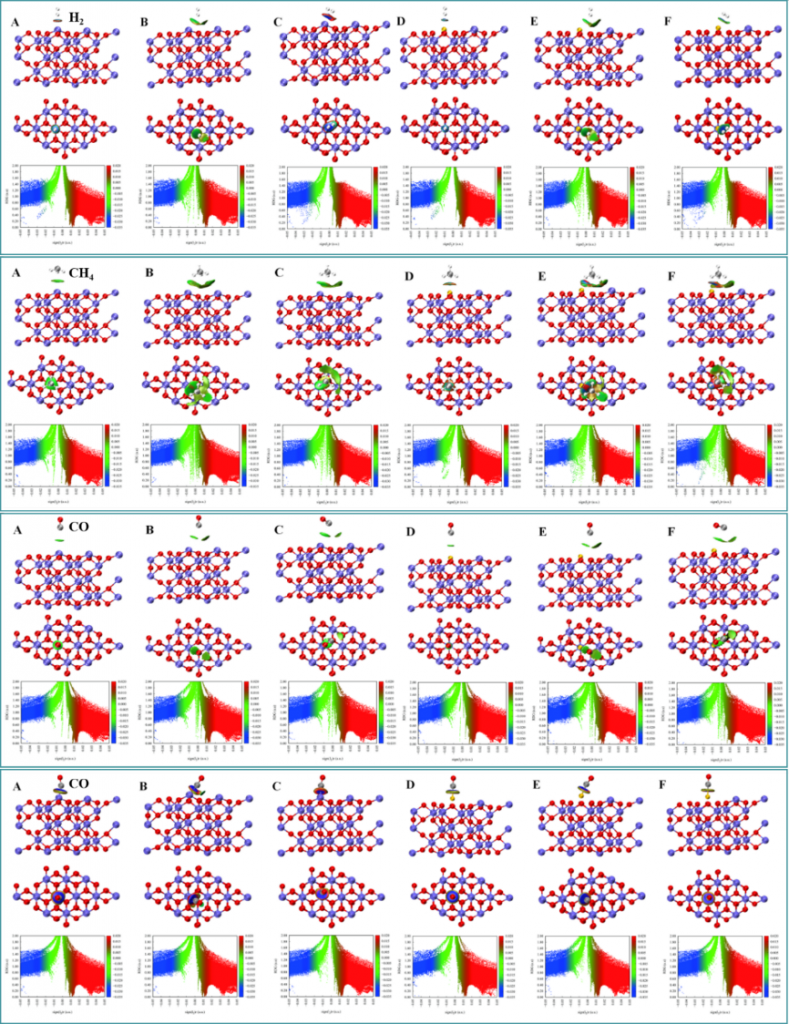
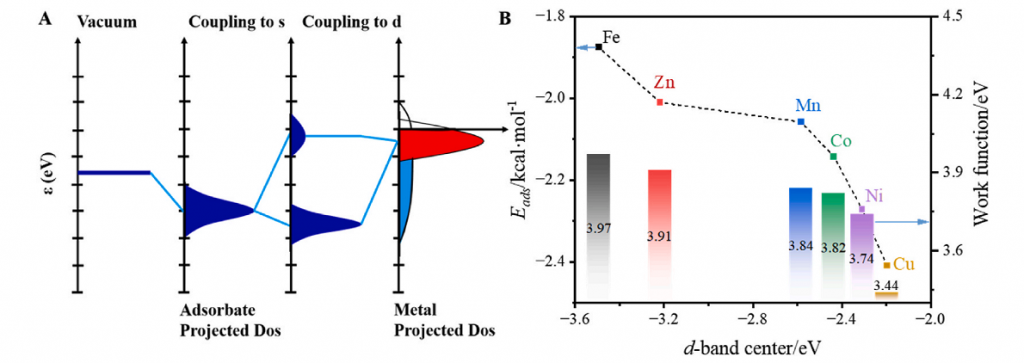
通过 RDG、IRI、CDD 和 ELF 方法实现了相互作用的可视化。RDG 与 IRI 分析显示相互作用强度顺序为 CO > CH4 > H2,与吸附能结果一致。CDD 定性展示了电子转移方向,并定量描述了转移量,直观呈现了吸附分子与氧载体间的非共价相互作用。基于态密度分析从电子结构层面阐释了吸附行为。过渡金属掺杂通过改变 TM…O 与 Fe…O 相互作用,有效调控载氧体电子结构,引起 d 带中心上移和功函数降低。其调控机制主要源于掺杂原子的电子结构差异、表面电荷失衡及晶格畸变效应。
总结
基于 DFT 计算,系统探究 H2、CH4 及 CO 在 Mn/Co/Ni/Cu/Zn 掺杂铁基载氧体表面的吸附机制。结果表明,H2 与 CO 最稳定的吸附位于桥位,CH4 优选空心位。EDA 揭示了主导作用力呈显著差异:H2 以轨道作用为主,CH4受静电作用主导,CO 物理吸附中轨道作用占比 45-57%,而化学吸附中轨道与静电作用共同主导。RDG 与 IRI 分析直观证实范德华力为主导的相互作用,其中Cu/ Ni/ Co掺杂体系表现出增强的吸附能力。电子结构分析表明,掺杂通过调控 TM···O/Fe···O 相互作用,诱导 d 带中心上移与功函数降低,其调控机制源于掺杂原子的独特电子结构、表面电荷失衡及晶格畸变效应的协同作用。
参考文献
- Jinpeng Zhang, Liangliang Meng, Huining Wan, Jieying Jing, Yurong He, Yuhua Wu, Jianbo Wu, Hui Zhang, Hongcun Bai, Decoding fuels and oxygen carriers interaction: Insights into site-specific adsorption mechanisms and driving forces of H2, CH4 and CO on doped iron-based oxygen carriers during chemical looping combustion, Journal of the Energy Institute, 2025, 123, 102326, DOI: 10.1016/j.joei.2025.102326