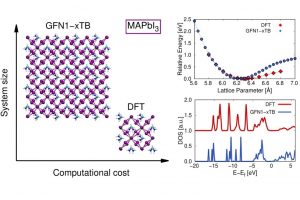
对金属卤化物钙钛矿材料性质的全面理解,对于提高未来太阳能电池的稳定性和效率至关重要。毫无疑问,计算模拟是在原子水平上描述金属卤化物钙钛矿的物理化学性质的一个非常有价值的工具。为了彻底了解这些复杂系统,需要对相对较大体系中的电子和离子进行同时建模,这对DFT计算来说代价很大。埃因霍温技术大学的研究人员,近期验证了最近的GFN1-XTB DFTB方法用于模拟卤化物钙钛矿性质的适用性。 他们评估了GFN1-xTB的性能,并计算了几种金属卤化物钙钛矿的结构、能量、振动和光电特性。对一系列具有不同化学成分(阳离子、金属、阴离子)以及结构相(立方、四方、正交)的卤化物钙钛矿进行了与实验、DFT计算的综合比较。 结果表明,GFN1-xTB方法可以处理比标准DFT计算更大的体系和更长的时间尺度,而计算成本很低,同时在计算各种结构和电子性质时保持较高的精度。虽然DFTB在模拟钙钛矿方面显示出很高的通用性,但仍有改进的余地,最显著的是某些结构相,经过几何弛豫后发生的结构畸变,以及含有复杂动态键的离子的错误电子描述。由于xTB哈密顿量的可调性质,作者设想通过重新参数化来改善DFTB的性能。基于AMS的ParAMS,能够训练ReaxFF、DFTB参数。 参考文献: J. M. Vicent-Luna, S. Apergi, S. Tao, Efficient Computation of Structural and Electronic Properties of Halide Perovskites Using Density Functional Tight Binding: GFN1-xTB Method, J. Chem. Inf. Model. 61, 4415 (2021)