
机器学习 DFT
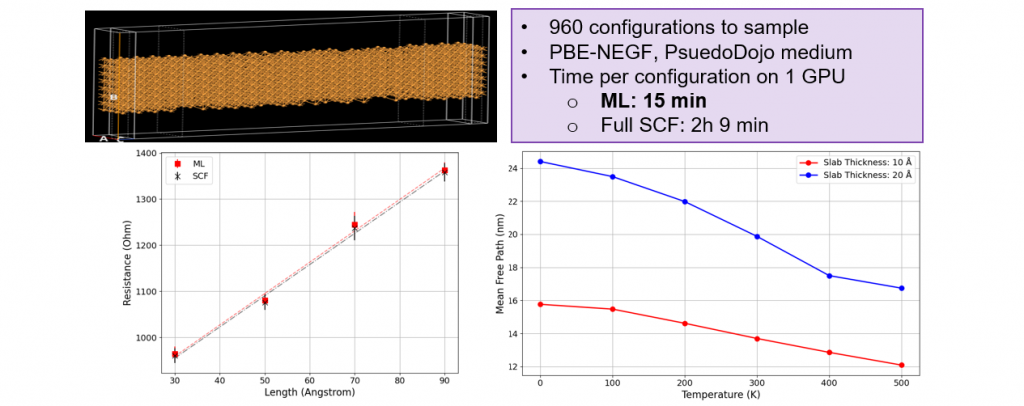
QuantumATK 本次发布的新版本引入了机器学习密度泛函理论(ML-DFT)。ML-DFT 模型经过训练可重现 DFT(GGA)级别的密度或有效势函数,能显著加速电子结构特性及输运过程的模拟流程。ML 训练得到的密度/有效势函数可用于:
- 作为标准 DFT 计算中自洽场循环的初始状态,大幅减少自洽场迭代次数;
- 直接预测 Hartree 差别势、总能量、能带结构、态密度、哈密顿量导数、透射等特性(无需完整自洽场循环)。通过替代传统 DFT 中最耗时的步骤(完整自洽场循环),ML-DFT 实现了 DFT 级计算速度的大幅提升,从而支持包含多达 1000-10000 个原子的大规模模拟,并助力以往传统 DFT 难以实现的高通量统计平均研究。
用户可以使用软件预先训练好的模型或自己针对特定体系训练模型。训练和使用模型都可以在图形界面中完成。
- 当前版本中包含的预先训练的模型:Materials Project 数据库中的密度、SiGe 密度、III-V 化合物密度、铜有效势、HKMG 多层堆叠分区密度等。
GPU 加速原子轨道基组 DFT 杂化泛函计算、平面波 DFT 计算和 G0W0 计算
在之前的 X-2025.06 版本中,QuantumATK 实现了 DFT- LCAO(LDA、GGA、metaGGA)以及半经验(SE)体相计算(包括能带结构、投影能带结构、投影态密度、布洛赫态)和器件(NEGF)计算的 GPU 加速功能。本次发布的 QuantumATK 版本新增了 DFT- LCAO 混合泛函、DFT 平面波方法、多体物理 G0W0 方法以及电子-声子耦合(EPC)计算的 GPU 加速支持。
- DFT-LCAO 杂化泛函 6x 加速
- DFT-PlaneWave 计算 10x 加速
- G0W0 计算 12x 加速
- 电声耦合(EPC)计算 6x 加速
- 平衡态(偏压为0时)的普通 DFT 和半经验加速比上一个版本也有提升
注:以上结果基于特定的体系和硬件测试,详情参见厂商新版发布说明文档。
半经验量子力学方法和器件 NEGF 方法改进
新版本为半经验分析计算(如投影态密度(DOS)与电子-声子耦合)以及基于半经验模型和密度泛函理论(DFT)的器件 NEGF 计算,带来了多项性能优化(包括加速和内存占用降低)。
- 采用不同半经验(SE)模型进行计算时的加速效果
- 投影态密度(PDOS)最高可达 4 倍;
- 电子-声子耦合最高可达 35 倍;
- 采用不同 SE 模型时,体力与应力计算最高可达 25 倍加速(通常仅占总模拟时间的很小部分)。
- 器件 NEGF 加速
- 使用多种 SE 模型进行 NEGF 计算时的加速效果。
- 器件自洽场最高可加速 3 倍
- 器件 DOS(正交模型,如最近邻模型)最高可加速 20 倍
- 透射计算最高可加速 1.5 至 3 倍。加速效果取决于器件的几何结构,宽器件越大,细长器件越小。
- DFT- LCAO 进行 NEGF 计算时的加速效果
- 能量与力计算最高可加速 12 倍
- 器件自洽场计算最高可加速 4 倍 + 收敛性更优
- 光电流计算最高可加速 7 倍
- 使用多种 SE 模型进行 NEGF 计算时的加速效果。
注:以上结果基于特定的体系和硬件测试,详情参见厂商新版发布说明文档。
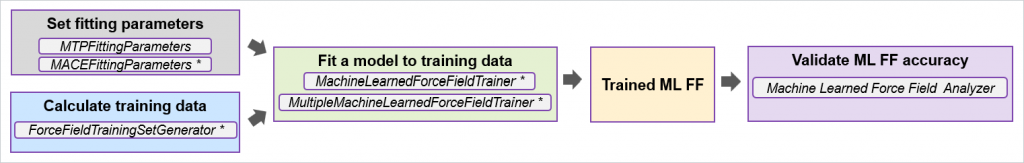
新一代模块化机器学习力场训练框架
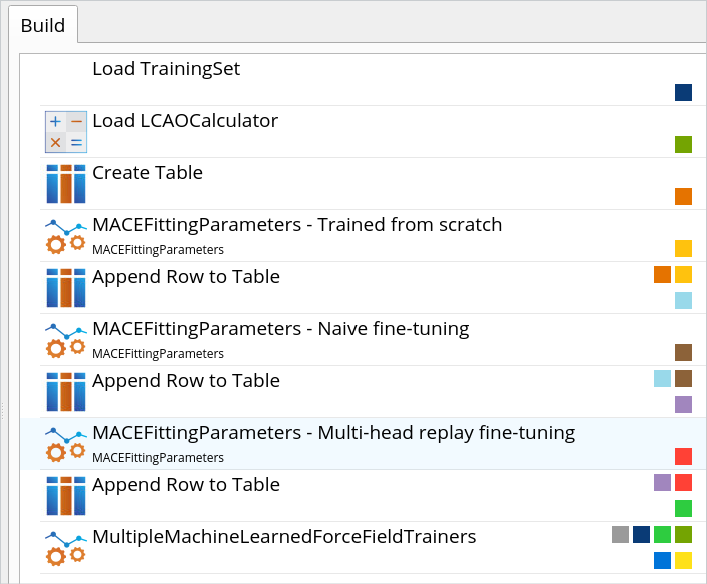
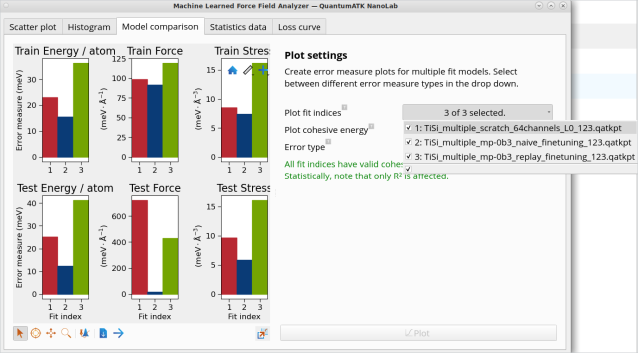
在之前的版本中,矩张量势(MTPs)与神经网络 MACE 机器学习力场(ML FFs)采用了不同的训练框架。本次版本通过引入通用模块化机器学习力场训练框架,显著提升了操作的便捷性。该框架包含数据生成、模型拟合与验证等独立模块,可适用于 MTPs、MACE 等多种机器学习力场模型的训练。新版特别新增专用工作流构建模块与机器学习力场分析工具,用户可通过 NanoLab 图形界面直观展示不同 ML FF 架构(MTP 与 MACE)的误差统计图,并通过单一图表对比多个模型拟合结果,快速获取整体模型性能评估。
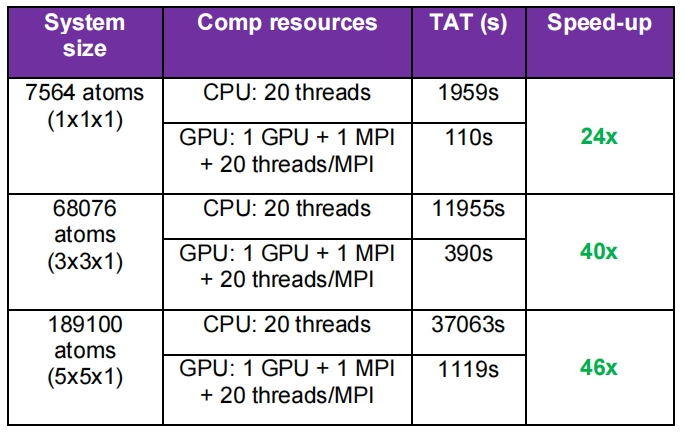
机器学习力场 GPU 加速比显著提高
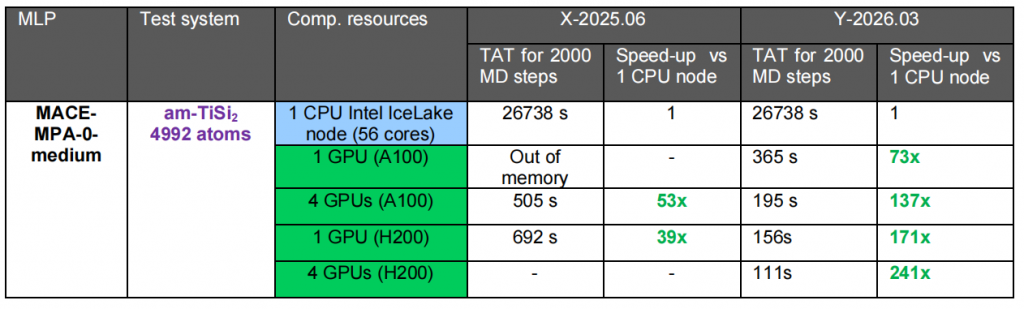
在之前的 X-2025.06 版本中,QuantumATK 为采用 MTPs 的离子动力学模拟引入了单 GPU 与多 GPU 加速技术,并为使用 MACE 和 MatterSim 神经网络势能的分子动力学模拟提供了多 GPU 加速方案。本次版本针对 MACE 和 SevenNet 神经网络势能实现了 GPU 性能的显著提升(3-5 倍),相较于 CPU 整体加速比最高可达 250 倍。
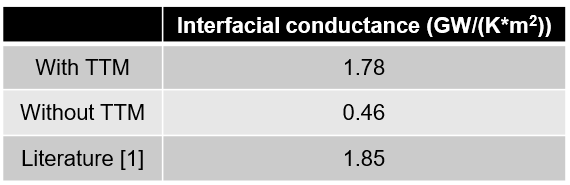
热学材料的精确模拟:MD 的双温模型
在典型的分子动力学(MD)模拟中,离子被视为与电子解耦(整个系统采用单一热库)。该方法提出了一种双温法(TTM),适用于包含电子-声子相互作用和电子阻滞效应的 MD 模拟。TTM 能够模拟含电子贡献的热输运(对含金属体系尤为重要),或非平衡电子加热(例如通过光-物质相互作用)。

全新的软物质动力学引擎
本次版本推出全新的 SoftMatterCalculator 工具,专为结合键合力场/有机力场的高 GPU 优化分子动力学与弛豫模拟设计,较 CPU 运算速度提升 24 至 50 倍。该工具适用于聚合物、流体及溶剂等软物质模拟场景,同时支持热塑性聚合物与热固性聚合物模型的构建。
- 在 NVE、NVT 和 NPT 系综中进行分子动力学(MD)模拟。
- 模拟应力-应变曲线、热传导、内聚能密度及分子扩散等物理特性。
- 加速构建真实聚合物模型,涵盖堆积、交联及弛豫/平衡阶段。
- 支持质心约束、原子约束与键约束。
- 支持脚本编程操作,同时可通过 CrosslinkBuilder 模块图形界面进行交联模拟。
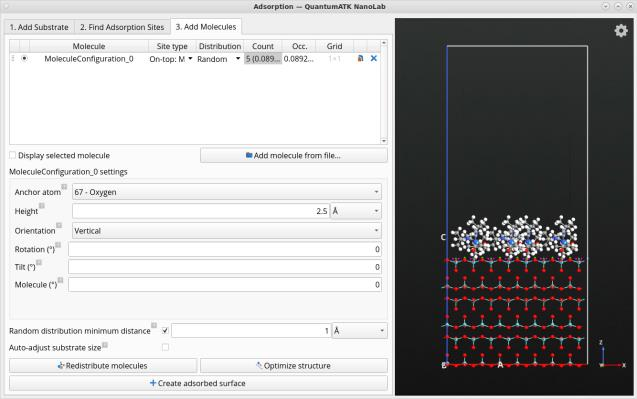
表面过程模拟改进
图形用户界面(GUI)中改进的吸附工具,用于生成吸附分子结构。
- 更友好的用户界面设计
- 直接在吸附工具中实现裂解与重复表面
- 自动化单元格拟合
- 通过刚体结构优化精炼结构(消除重叠、原子碰撞等)
- 改善分子表面取向的功能模块
- 使用元素符号标记吸附位点的改进方法
SurfaceProcessSimulation 动力学模拟新增两个函数(同样位于 GUI 的 HookFunctions 模块):
- AugerNeutralizationHook:当分子接近表面时解除其约束,用于模拟 Auger 中和过程;
- ElectronicStoppingHook:对分子中的元素施加阻力。
其他动力学计算改进
- AKMC 中的 Iso-ARTn 鞍点搜索方法
- 提高了寻找过渡态的成功率,并减少了 40% 的力评估次数。
- 将外推密度(来自前三个步骤)作为分子动力学和几何优化的初始猜测值
- 支持 GGA 和 MGGA 泛函
- 系统性地改进并减少自洽场步骤数量(默认设置)
其他新功能和增强
- 升级 Materials Project 数据库接口
- 支持 Materials Project 数据库最新 API规范
- 包含近 2-3 年新增材料数据,神经网络模型训练框架用于预测材料通用特性
- 采用机器学习材料性质训练器(MachineLearnedPropertyTrainer)对原子构型进行性质预测训练。
- 与仅能学习构型能量及能量导数属性(如力场和应力)的力场训练不同,性质预测侧重于学习构型层面或原子级特性,例如带隙、形成能或原子电荷。
- 可从零开始训练,也可基于 MACE 等预训练基础模型进行微调。
- 支持脚本化操作。