
η²-C₂B₁₀H₁₀及其衍生物与第Ⅹ族过渡金属的相互作用
碳硼烷是一类独特的二十面体硼碳簇合物,具有优异的热稳定性、疏水性和生物惰性,使其在药物递送、有机金属化学等广泛领域备受关注。其脱氢衍生物邻位碳硼烯可作为多功能配体,以 η1 或 η2 模式结合过渡金属,由此在早期和晚期金属中形成结构明确的碳硼烷基 / η2-碳硼烯配合物。虽然过渡金属-配体相互作用传统上由 Dewar–Chatt–Duncanson (DCD) 模型描述,但对于芳炔和 C₆₀ 配体,已有报道其成键方式偏离了传统模型,表现为配位键与电子共享键共存的(D+E)相互作用模式。受这些先例启发,作者采用自然键轨道(NBO)、量子理论原子(QTAIM)、电子局域化函数(ELF)和能量分解分析-自然轨道化学价(EDA-NOCV)等方法研究了第 10 族过渡金属配合物 TM–(η2-1,2-C2B10H10–nXn)(TM = Ni, Pd, Pt; n = 0–2; X = I, Br, Ph)中的成键。这些研究表明,与遵循 DCD 模型的炔/芳炔类似物不同,邻位碳硼烯配合物也采用了配位键+电子共享组合的成键模式,通过基于 AMS 软件中的 EDA-NOCV 方法分析发现,其中过渡金属片段作为阳离子片段,与阴离子型邻位碳硼烯片段相互作用,并通过占主导地位的静电相互作用,辅以 π 共享作用形成稳定化学键。该工作凸显了邻位碳硼烯独特的成键适应性,并确立了其作为超越经典模型范畴的电子柔性配体的地位。( Inorg. Chem. 2025, DOI: 10.1021/acs.inorgchem.5c04380)
高自旋钴基与镍基金属化硼烯配合物[(OC)BM(CO)ₙ][M₂(CO)₆]⁺ (M = Ni, Co; n = 2, 3) 的结构、成键及稳定机制研究
硼炔(:BR)因其独特的两性反应活性,在小分子活化领域引起了广泛关注,因此稳定这类物种对于实现其实际应用至关重要。近年来,金属化硼炔(:BM)作为有机硼炔的新变体出现,但其稳定机制尚不完全清楚。本研究在气相中生成了阳离子型钴基与镍基金属化硼炔,利用红外光解离光谱和密度泛函理论计算对其饱和羰基配合物进行了结构表征,揭示了其高自旋电子态以及具有C₂v对称性的平面几何构型。结合 EDA-NOCV 分析发现,这些物种中的硼中心表现出显著的 σ 酸性和 π 反馈能力,其羰基活化程度与碱稳定的硼炔相当。电子结构和化学成键分析阐明了不饱和硼中心与周围金属原子之间的相互稳定机制,证实了构建两性金属化硼炔的策略可以扩展至单价金属之外。(Inorg. Chem. 2025, DOI: 10.1021/acs.inorgchem.5c03853)
单环 E₆H₆ 与 E₄H₄(E = C, Si)的结构与芳香性
本文报道了对单环分子 E₆H₆ 和 E₄H₄(E = C, Si)的结构与芳香性进行的量子化学密度泛函计算。结果表明,碳同系物与硅同系物之间存在显著差异。苯(1b)是C₆H₆势能面上的全局能量最小值,而平面D₆h对称的Si₆H₆(2d)并非能量极小点,且D₃d构型2c的能量高于其棱柱烷异构体2a。已知的稳定苯基化合物数量众多,但实验上唯一已知的Si₆R₆化合物具有三环结构2b,其能量低于2c。形成鲜明对比的是,碳的同系物异构体1c的能量比1b高出超过120 kcal mol⁻¹。碳化合物C₆H₆和C₄H₄表现出特征性的反应偏好:苯(1b)倾向于取代反应,而环丁二烯(3a)倾向于加成反应。硅同系物Si₆H₆(2c)对取代反应的偏好弱于苯,但四硅环丁二烯(4a)同样偏好取代反应而非加成反应。
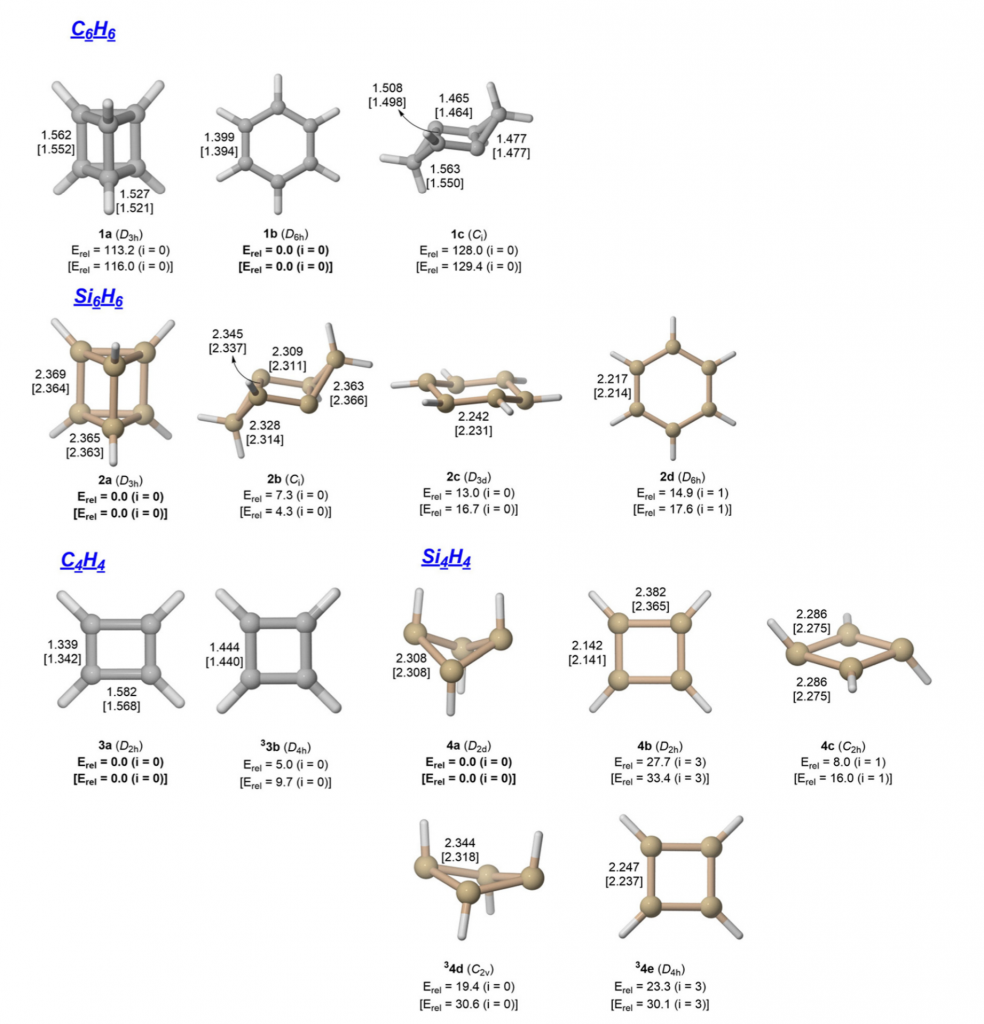
对环状化合物与非环状参比体系的(伪)π 共轭体系,进行了 EDA-NOCV 计算比较,表明碳体系存在芳香稳定化/反芳香 destabilization 效应。硅体系的计算结果则无法得出明确结论,且由于部分硅体系严重偏离平面构型,σ 和 π 相互作用的分离变得困难。核独立化学位移值(NICS)并非衡量 π 共轭所致芳香稳定化的可靠指标。针对周期表第一周期元素化合物建立和发展的化学成键模型,对于含有较重主族原子的分子仅具有有限的适用性。其根源在于原子 s/p 价层轨道的半径:第一周期元素的 s 和 p 轨道半径非常接近,从而形成有效的 sp 杂化。而较重原子的化学键具有高得多的 p 特征,因其 p 轨道半径大于价层 s 轨道的半径。(Phys. Chem. Chem. Phys., 2025, DOI: 10.1039/D5CP02697K)
打破禁区,s区-镧系金属成键配合物
镧系金属由于其 4f 轨道高度屏蔽、径向扩展性弱,难以与其他原子轨道有效重叠,长期被视为不参与共价成键的“惰性”金属。在多数配合物中,镧系元素仅通过静电作用与配体中的 O 或 N 等硬碱相互作用,呈现典型的离子型键合行为。相比之下,镧系–金属之间的直接金属–金属键(Ln–M)极为罕见,已知例子多限于 Ln–p 区金属(如 Sn、Ga、Al)体系,且多依赖阴离子桥连或范德华相互作用。镧系–s区金属(如Mg、Ca)之间的共价成键此前从未被实验证实,主要因两者都电正性极强、缺乏成键轨道匹配。
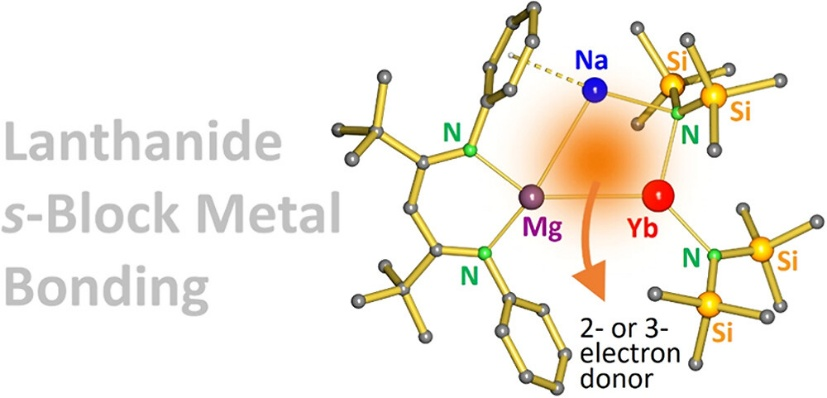
该项工作不仅首次构建了 s 区–镧系金属键(Mg–Yb),还系统比较了与同周期 Ca 的结构与电子差异,通过 EDA-NOCV 等高水平理论工具揭示其成键本质,是对金属键图谱的关键补充,也为未来设计新型镧系–主族催化体系奠定了基础。(J. Am. Chem. Soc., 2025, 147(15): 12555-12561. DOI: 10.1021/jacs.4c17853)