
导语
较重主族元素的低氧化态氢化物因极度不稳定而长期被视为“难以捉摸”的研究难题,然而,Oriol Planas 教授团队近期在《Journal of the American Chemical Society》发表的题为《A Crystalline In(II) Hydride》的突破性成果,成功打破了这一僵局。该研究首次报道了首例稳定且具有晶体结构的低氧化态铟氢化物,通过双(N-杂环卡宾)基硼酸配体骨架,巧妙利用空间位阻设计、非共价相互作用与两性离子电荷分布的协同作用,实现了对这种低价重主族物种的有效稳定。研究表明,该化合物不仅拥有强烈的共价 In–In 相互作用和高度极化的 In–H 键,更展现出反常的亲核特性,能够参与卤化、烷基化、氢化脱氟及硫族解等丰富反应,且在整个过程中 In–In 键始终完好无损。这项工作不仅揭示了赋予此类稀有化合物非凡稳定性与反应性的核心设计原理,将较重第 13 族氢化物重新定义为小分子官能化的强力平台,更为主族氢化物化学拓展至“曾被认为不可及”的低氧化态领域奠定了坚实的概念与结构基础。
研究背景
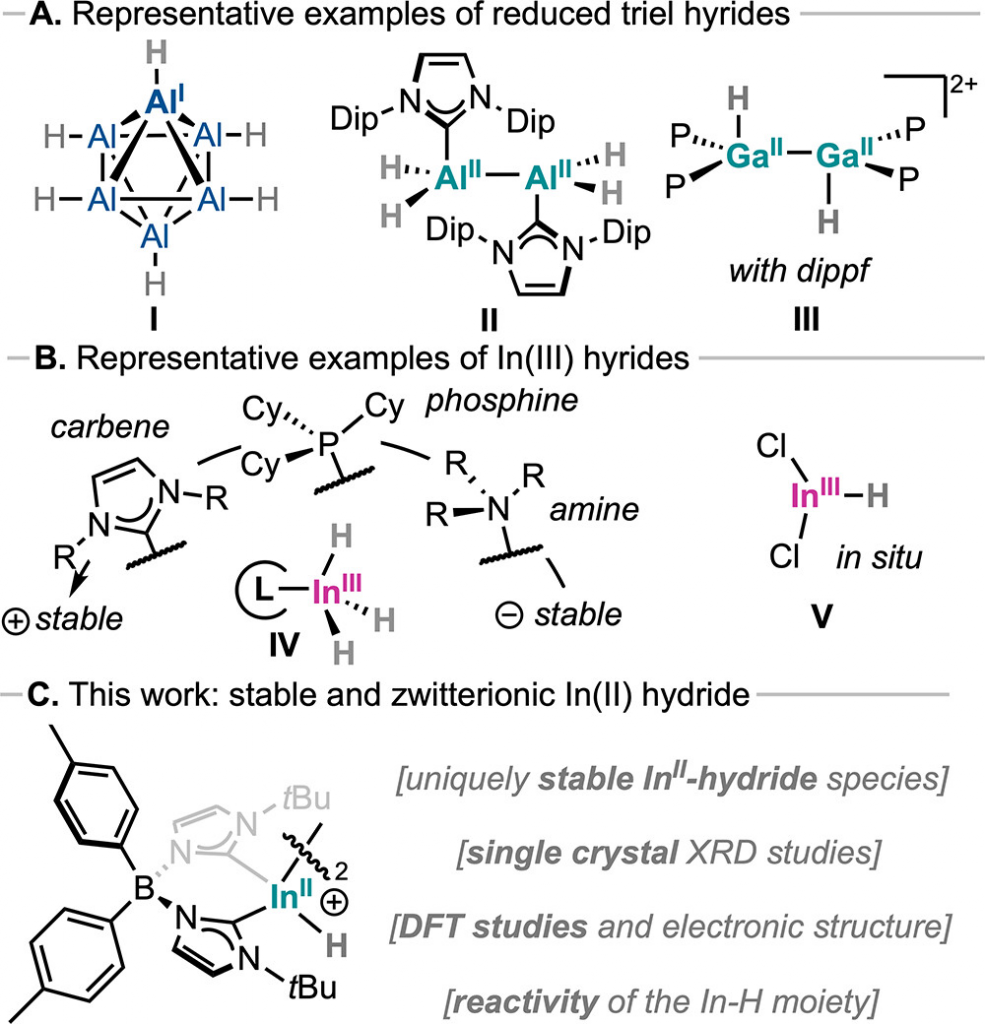
氢化物作为支撑加氢、还原及小分子活化等核心过程的关键中间体,其研究领域正经历从传统过渡金属向主族金属的重要范式转变。近年来,第 14、15 族以及第 13 族的铝、镓已相继突破常规,成功实现了低氧化态氢化物的分离(图 1A);然而,同属第13族的较重元素铟和铊却严重“掉队”。尽管高价 In(III) 氢化物已被成功表征,但低氧化态铟氢化物因极易经由形成 In-H-In 桥并释放氢气而分解为铟黑,长期仅能在气相或极低温下被瞬态观测,导致“此类物种不可及”成为领域内的刻板印象。理论上,这类低价重主族氢化物本应具备高度极化的键和独特的氧化还原灵活性,但“分离不出来”的瓶颈让一切潜在的反应模式都无从评估。受近期卡宾化学启发,Oriol Planas 教授团队巧妙引入大位阻、阴离子型的双 (NHC) 硼酸配体(图1 C),凭借其强给电子性质与显著的空间位阻,精准靶向并克服了低价铟氢化物的失稳难题,从而为最终实现首例低于 +3 氧化态的稳定晶体铟氢化物的合成奠定了坚实的基础。
研究内容
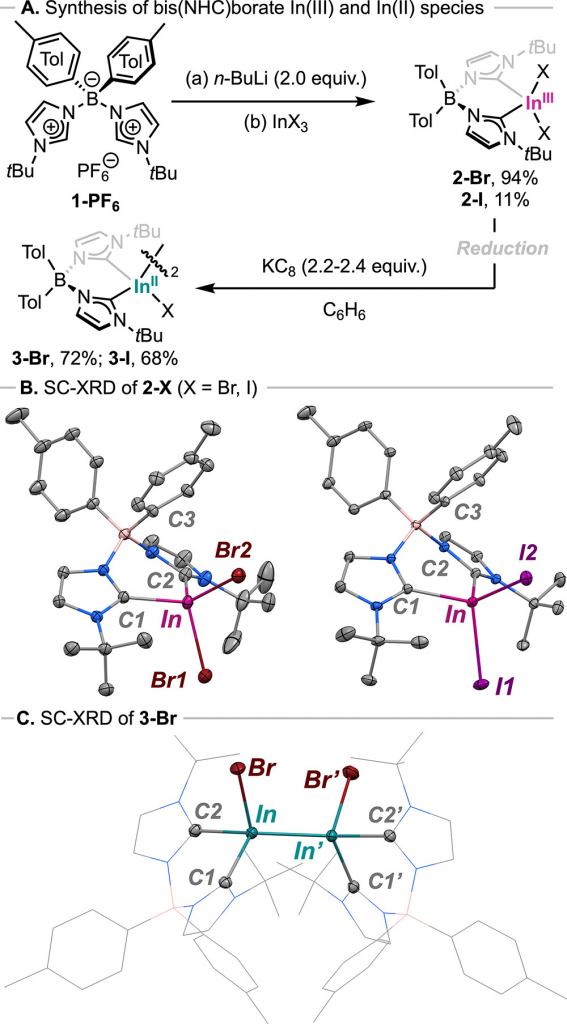
通过逐步还原与取代策略实现了低价铟氢化物的合成(图 2A)。以双 (NHC) 硼酸配体为骨架,研究首先合成了 In(III) 卤化物配合物 2-Br,单晶分析表明其中心铟呈畸变四面体配位(图 2B),且配体与金属间存在弱相互作用。随后,使用 2.2 当量 KC8 在苯中还原 2-Br,以 72% 的产率获得了含 In-In 键的 In(II) 二聚体物种 3-Br(图 2C)。在确认该二聚体具有良好的稳定性后,进一步探索了其与氢化物试剂的反应,在筛选了 KH、LiAlH4 等多种试剂后,发现使用 K-Selectride 在干燥乙醚中处理 3-Br,能够成功实现卤素-氢交换,最终以 30% 的产率分离出目标二氢化物物种 3-H(图 3A)。
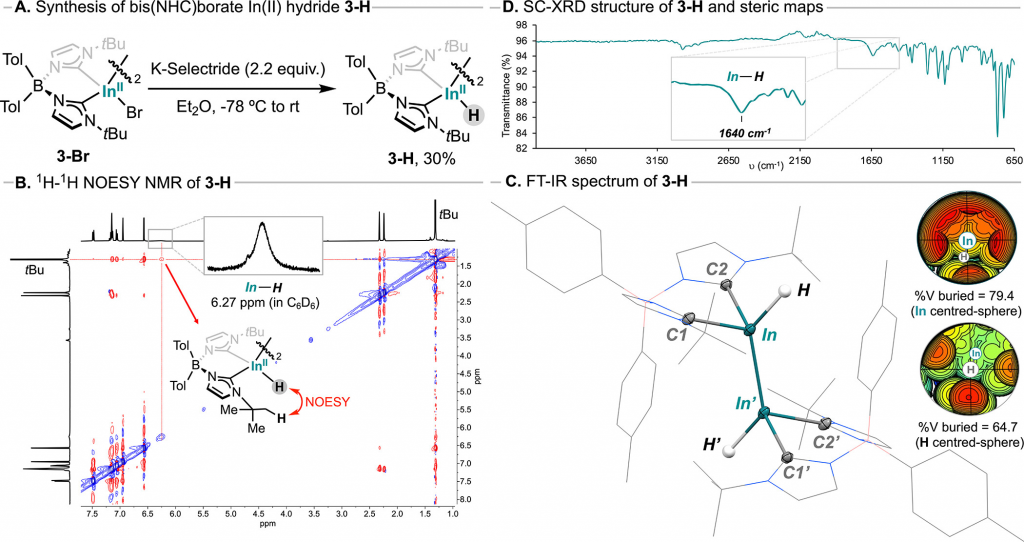
对目标产物 3-H 通过系统的谱学、晶体学及计算手段明确了其结构与性质。溶液 NMR 研究表明(图 3B),3-H 在 C6D6中于 δH = 6.27 ppm 处表现出对应 In-H 键的宽单峰,结合 NOESY 和 DOSY 谱图数据以及变温 NMR 结果,证实该物种在溶液中为稳定的二聚体结构,不存在单体-二聚体动态平衡。固态表征方面,FT-IR 光谱在 1640 cm-1处观测到特征性的 ν(In–H) 伸缩振动带(图3C);单晶X射线衍射分析(图3D)不仅确认了 In-In 键的存在及 1.75(2) Å 的端基 In-H 键长,并且在晶体结构中还明显存在涉及 tBu 取代基的分子内单聚体间 In(II)–H···H–C 短距离接触(dH–H < 2.4 Å)。该化合物在固态和溶液中均具有良好的空气稳定性,暴露数小时后仍能保持其完整性,并在高温下保持稳定数小时。此外,计算化学研究表明,3-H 消除 H2 的活化能垒高达 48.6 kcal/mol,结合对配体空间位阻(中心铟屏蔽率达 79.4%)从机制上阐明了该低价 In(II)-H 二聚体在固态及溶液中表现出异常热稳定性和一定耐空气能力的根本原因(图3D)。
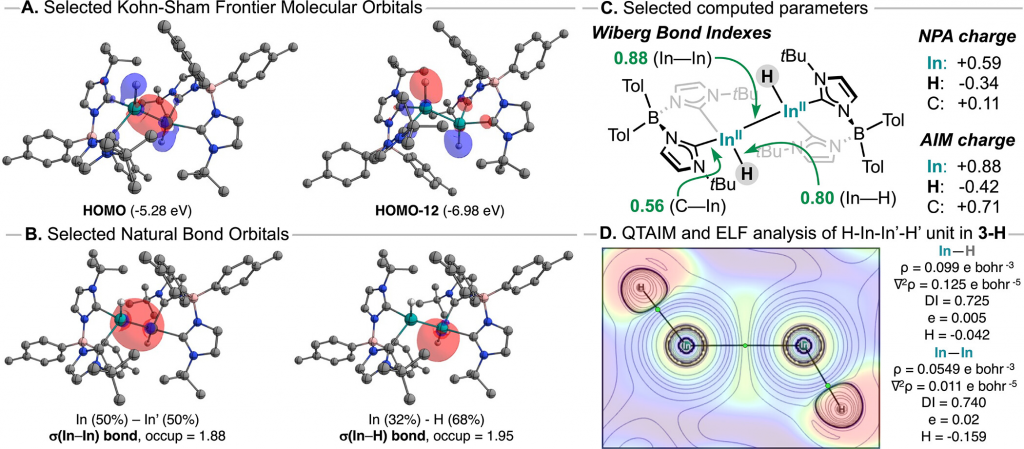
通过在 PBE0-D4/def2-TZVPP 水平下的理论计算,研究系统解析了 3-H 的电子结构。分子轨道分析表明,其 HOMO 主要对应于 In–In 键并部分离域至端基氢化物配体(图 4A)。自然键轨道(NBO)与 Wiberg 键级(WBI)分析证实,In–In 键(0.88)和 In–H 键(0.80)均表现为单键特征,而 In–Ccarbene 键(0.56)则呈现高度极化的配位键性质(图 4B)。自然布居分析(NPA)显示,In 中心带有显著的正电荷(+0.59e),而氢化物配体带有负电荷(-0.34e),表明其具有亲核特征(图 4C)。进一步结合电子密度拓扑(QTAIM)与电子局域函数(ELF)分析(图 4D),研究定量刻画了分子内的键型差异:In–H 为典型的极性共价键;In–C 键存在共价稳定化作用但电子共享度较低;而 In–In 键的拉普拉斯值显示其电子积聚较少,但 ELF 分析给出了明确的双中心盆描述,结合相对较大的局部电子密度 H 负,表明存在金属-金属相互作用。
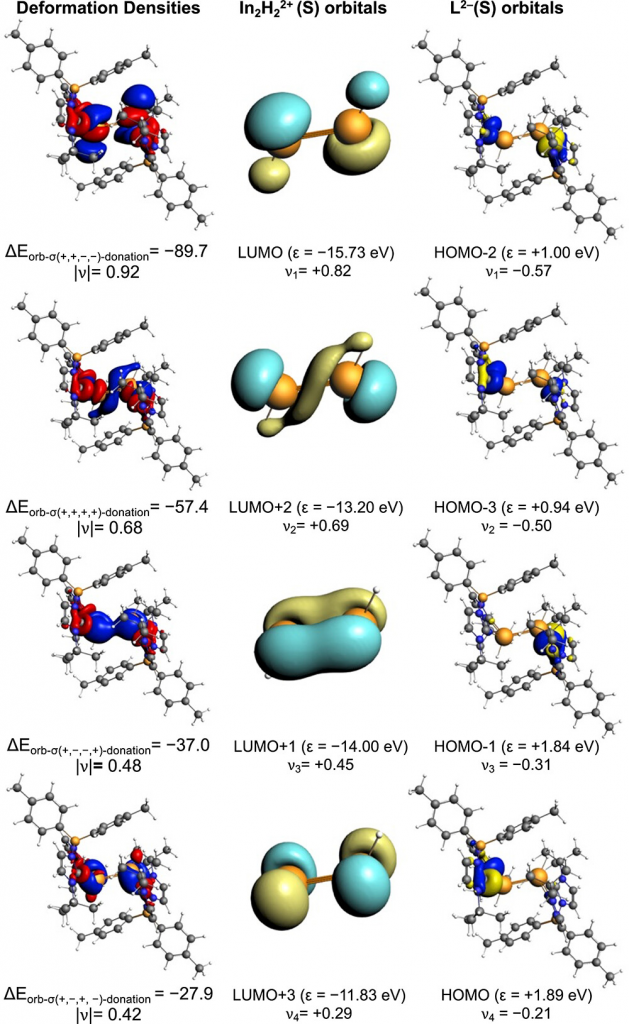
为量化阐明 3-H 的稳定性来源,研究采用 AMS 软件中 ADF 模块的能量分解分析(EDA)结合化学价自然轨道(NOCV)方法,将体系拆分为 [In2H2]2+ 核心与配体 [L]2- 片段,并与经典的 β-二亚胺类似物进行了严格对比。EDA 结果显示(表1),3-H 中核心与配体的总相互作用能高达 -648.1 kcal/mol,较 NacNac 类似物(-582.2 kcal/mol)高出约 66 kcal/mol。尽管 3-H 具有更长的给体-受体距离(3-H 中 In-C 为 2.24 Å,而 NacNac2In2H2 中 In-N 为 2.20 Å)和更大的 Pauli 排斥(3-H:ΔEPauli = 410.2 kcal/mol;NacNac2In2H2:ΔEPauli = 306.0 kcal/mol),但其绝对静电贡献(高出107 kcal/mol)和轨道贡献(高出54.4 kcal/mol)均大幅增加。NOCV 分析(图5)进一步将共价成分溯源为配体孤对电子向 [In2H2]2+ 空轨道的四通道 σ-给电子作用,双 (NHC) 硼酸配体的整体给体强度经计算为 −210.0 kcal/mol,显著强于 NacNac 配体(−178.6 kcal/mol)。这些量化数据表明,双 (NHC) 硼酸配体框架通过优化的强给电子能力与静电相互作用,高效稳定了高活性的低价态 [In2H2]2+ 核心,这是实现该低氧化态 In(II)–H 物种稳定存在的关键因素。

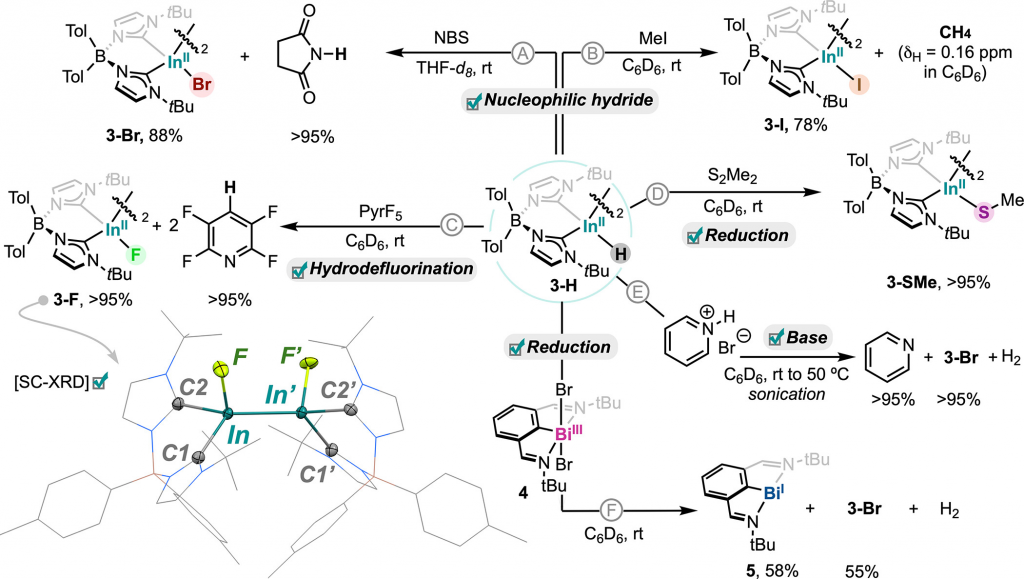
基于计算预测,研究对 3-H 进行了一系列反应测试(图6)。结果表明,3-H 与 NBS、碘甲烷反应直接证实了 In–H 键的亲核取代机制;与五氟吡啶反应则高产率生成了罕见的低价铟氟物种。此外,3-H 还能发生硫醚化、酸碱质子化及将 Bi(III) 还原为零价铟宾等反应。尤为关键的是,在所有过程中,其独特的二聚体骨架与 In–In 键始终保持完好,确立了 3-H 作为真正亲核性铟氢化合物的身份。
总结与展望
Oriol Planas 教授团队成功合成了首例兼具稳定性与结晶性的低价态铟氢化物 3-H,突破了重主族元素氢化物因易分解析氢而难以捕获的瓶颈。研究表明,双氮杂环卡宾硼酸盐配体通过空间位阻保护、非共价相互作用以及两性离子电荷分布的协同作用,有效稳定了分子内的共价 In-In 键与高度极化的 In-H 键。基于这种特殊的电子结构,3-H 表现出明确的亲核性特征,能够在完好保留 In-In 金属键的前提下,参与卤化、烷基化、脱氟、硫解及还原等多种化学反应。该工作不仅从实验和计算层面揭示了稳定低价重主族氢化物的设计原则,确证了低于 +3 氧化态铟氢化物化学的可行性,同时也将重 13 族氢化物重新定义为小分子活化与转化的可靠平台,为向传统上被认为难以触及的低氧化态主族氢化物领域的延伸奠定了结构与概念基础。
参考文献
- Mouriki O, Tizzard G J, Coles S J, et al. A Crystalline In (II) Hydride[J]. Journal of the American Chemical Society, 2026, 148(5): 5783-5792. DOI: 10.1021/jacs.5c22490
感谢胡钧员老师供稿!