
导语
在传统的化学认知中,氮气(N2)常被视为一种结合较弱、惰性极强的配体。然而,由 Gernot Frenking 教授团队近期发表于《Chemical Science》上的研究论文 Dinitrogen complexes N2L2(L = N2, CO, CS, NO+, CN−) 挑战了这一认知。研究证明,在特定的电子激发态下,中心的 N2 单元能展现出强烈的“吸电子”特性,即作为双路易斯酸与 CO、CS 等配体形成稳定的化学键。通过 AMS 中 ADF 模块的 EDA-NOCV(能量分解分析结合自然化学价轨道)这一计算工具,研究进一步揭示了这类分子的稳定性主要源于配体向中心 N2 的单向电荷转移,而非传统的反馈 π 键作用。这一成果不仅深化了对氮气化学性质的理论理解,也为新型高能氮材料的合成路径提供了重要的科学依据。
研究背景
自 1964 年三苯基膦嗪被发现以来,N2L2 类分子的真实成键本质长期存在争议,直到后续X射线衍射确证其具有反式构型与异常偏长的 N–N 键,理论分析才揭示其中心 N2 处于高激发态并与配体形成 L→(N2)←L 的强配位键。近期,研究团队在低温下成功合成了新型氮同素异形体 N6,尽管其被广泛视作 N₃ 的二聚体,但 N6 极度相似的反式构型与长 N–N 键提示:它可能属于 N2L2 化合物类,实质上是中心高激发态 N2 与两个末端 N2 发生的配位作用。基于这一新视角,Gernot Frenking 教授团队采用高精度量子化学方法重新解析了 N6 的电子结构与解离机制并系统拓展计算了等电子体系 N2L2(L = CO, CS, NO⁺, CN⁻)的几何构型、键解离能与振动光谱,旨在澄清该类分子的成键争议,为未来实验合成与表征提供理论指导。
研究内容
几何结构优化表明,所有 N2L2(L = N2、CO、CS、NO+、CN⁻)的平衡构型均保持 C2h 对称性,配体 L 围绕中心 N2 单元呈反式共面(trans-periplanar)排列(图1)。其中,中心 N–N 键长表现出显著的配体依赖性:当 L 为 N2 或 CN⁻ 时,键长介于 1.450–1.488 Å,呈现典型的单键特征;而当 L 为 CO、CS 或 NO⁺ 时,键长显著缩短至 1.355–1.386 Å,则表现出明显的双键特性。
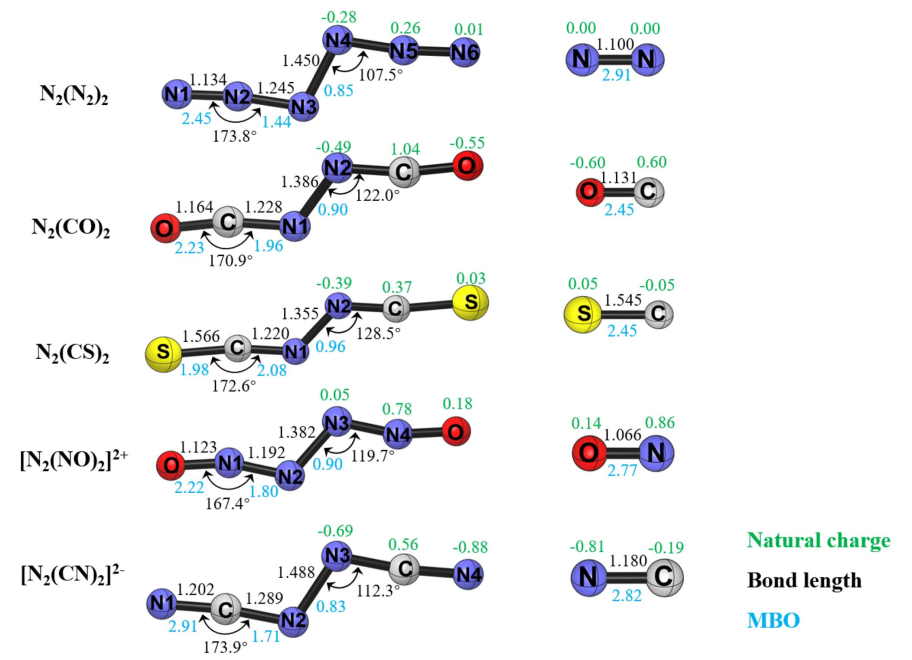
热力学分析表明,除 N2(CS)2(ΔG = 3.6 kcal mol⁻¹)在室温下稳定外,其余配合物均倾向发生异裂解(N2L2 → N2 + 2L,不稳定程度排序:L = NO+ > N2> CN- > CO),且异裂解在热力学与动力学上均绝对优于中心 N–N 键的均裂解(表1)。
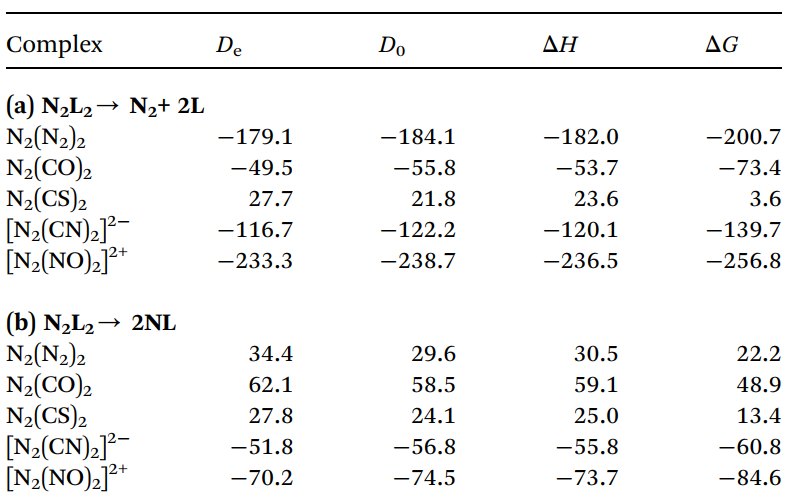
如图 2 所示计算表明,异裂解路径占据绝对主导地位,其活化自由能垒(ΔG⁺)在 16.9 kcal mol⁻¹(L = N2)至 84.2 kcal mol⁻¹(L = CS)之间变化()。在 N₆ 分子中,断裂键级更高、键长更短的侧翼 (N2)–(N2)2 键的能垒,反而显著低于断裂中心 N3–N3 键的能垒(后者均裂能垒高达 26.1 kcal mol⁻¹)。这一结果表明,断键所需能量不仅取决于键本身的强度,还取决于断裂过程中碎片电子结构的重排代价。该反常现象有力支持了N2→(N2)←N2 的给体-受体成键模型,合理解释了 N6 直接解离为 3 个 N2 的实验事实。
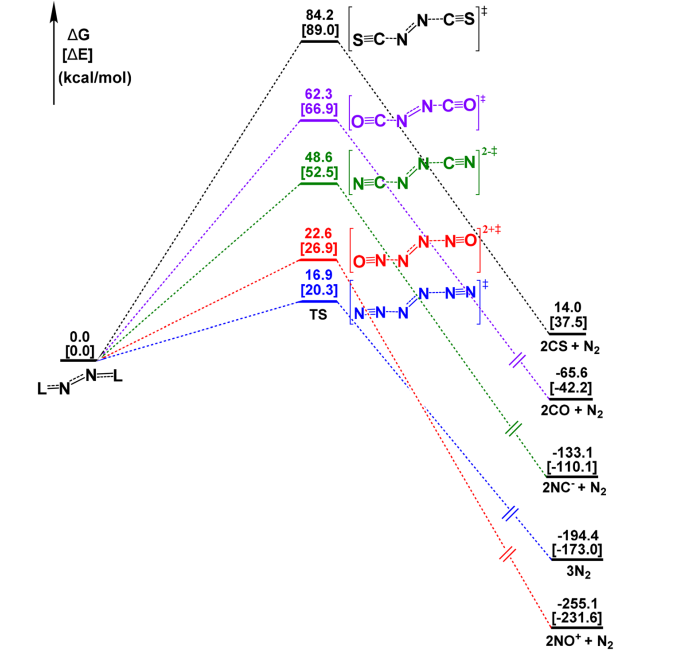
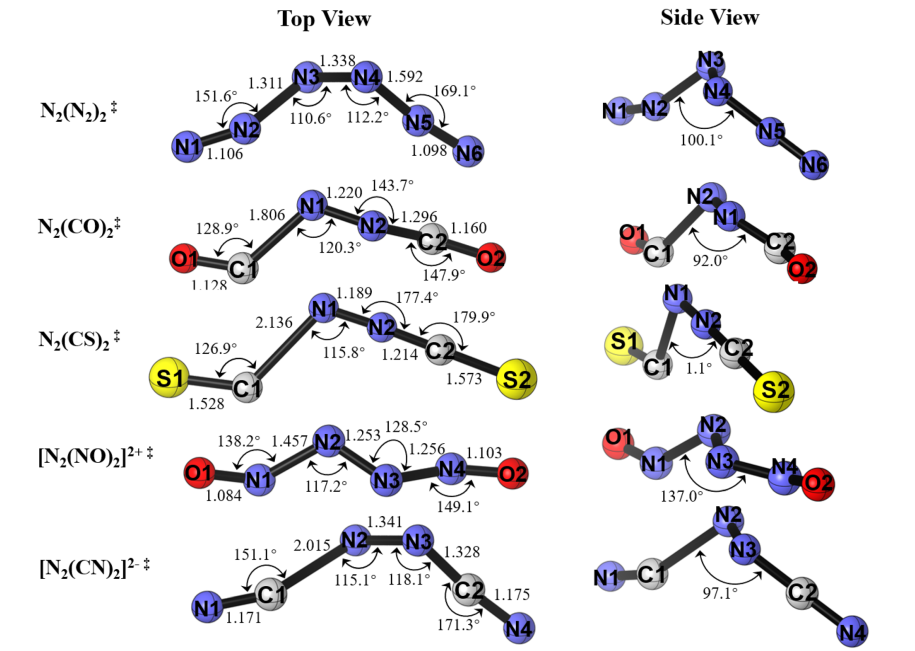
过渡态与 IRC 计算进一步证实(图 3),该异裂解过程是协同但高度异步的,两个 N2–L 键依次断裂,且在过渡态中中心 N–N 键反而缩短,进一步印证了中心 N2 单元在解离过程中的核心受体角色。除 N2 (CS) 2 的过渡态呈现近线性结构且伴随一个配体键显著拉长外,其余 [N2L2]≠ 过渡态均表现为配体呈顺式排列的非平面交错构型。
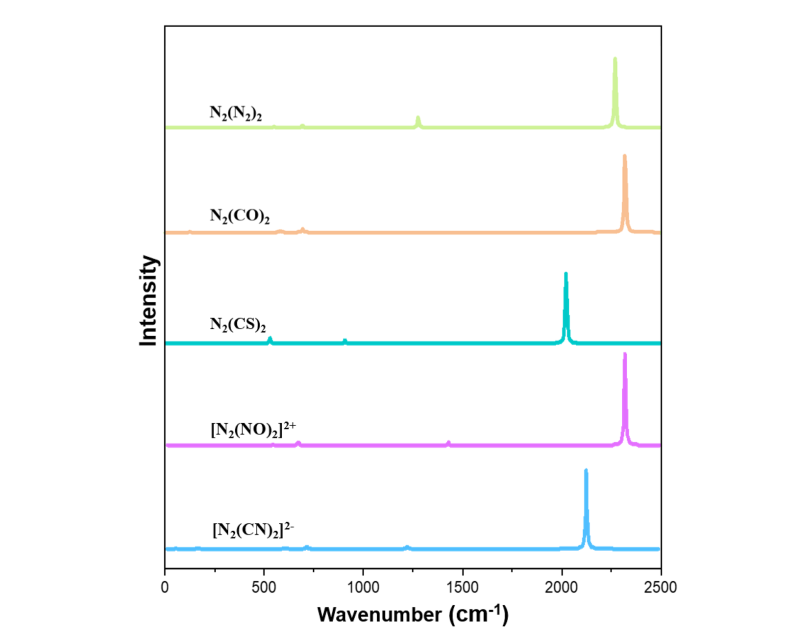
通过对 N2L2 系列分子的红外光谱的研究发现,这类化合物的红外光谱具有高度一致的特征,即末端配体的不对称伸缩振动(asymmetric stretching mode,νas)呈现最强吸收信号,而对称伸缩振动(symmetric stretching mode,νs)为红外中不活跃(图 4)。以 N6 为基准,该特征峰在 L = CO 和 NO⁺ 时发生蓝移,在 L = CS 和 CN⁻ 时发生显著红移,这一规律为分子的光谱鉴定提供了关键依据。
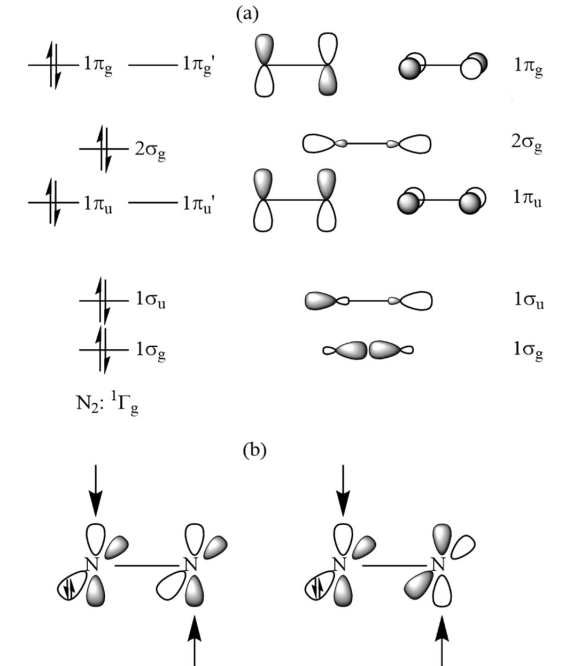
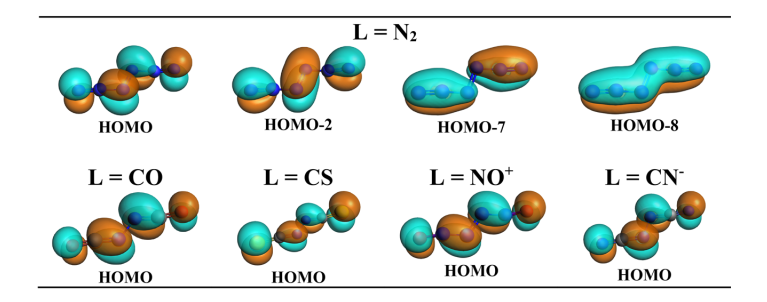
分子轨道分析为“中心 N2 以高激发态 (1)¹Γg 参与成键”的假设提供了决定性证据。计算准确识别出配体向中心 N2 空轨道注入电子的面内 σ 轨道相互作用;同时,体系中确实存在 4 个占据的价层面外 π 轨道(节点数依次递增),而非常规的 3 个,完全符合 (1)¹Γg 态双占据特征的预期(图 5)。此外,所有分子的 HOMO 均表现为中心 N₂ 的面外 π* 轨道与配体 π* 轨道的混合(图 6),进一步确立了该特殊的成键机制。
此外,通过能量分解分析-自然轨道化学价键(EDA-NOCV)方法对 N2L2 系列分子片段间相互作用的深入计算,进一步定量揭示了其复杂的成键本质与能量贡献机制。对于 N6 分子,计算表明其最优成键描述为中心处于激发单重态的 N2 与两个基态末端 N2 之间的配位作用(N2→(N2)←N2)(表 2)。进一步的轨道相互作用分解显示,这种配位作用以 σ 给予绝对主导(占总共价作用的 75%),而 π 反馈极弱(仅占 17%)。σ 电子进入中心 N2 的成键空轨道,合理地解释了配合物中心 N–N 键相比自由激发态显著缩短的现象(图 7)。
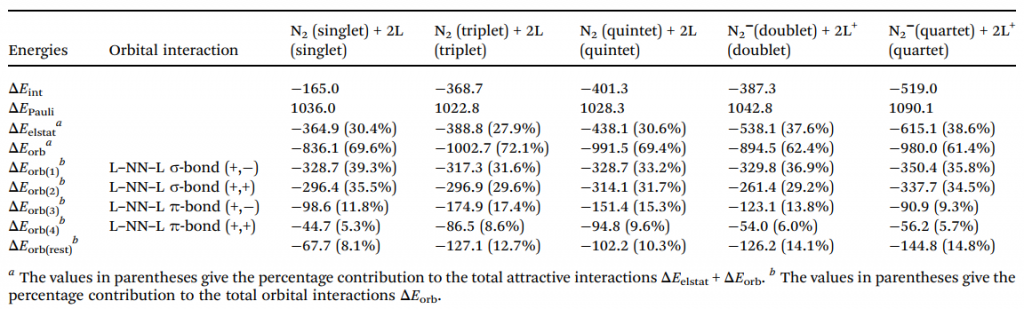
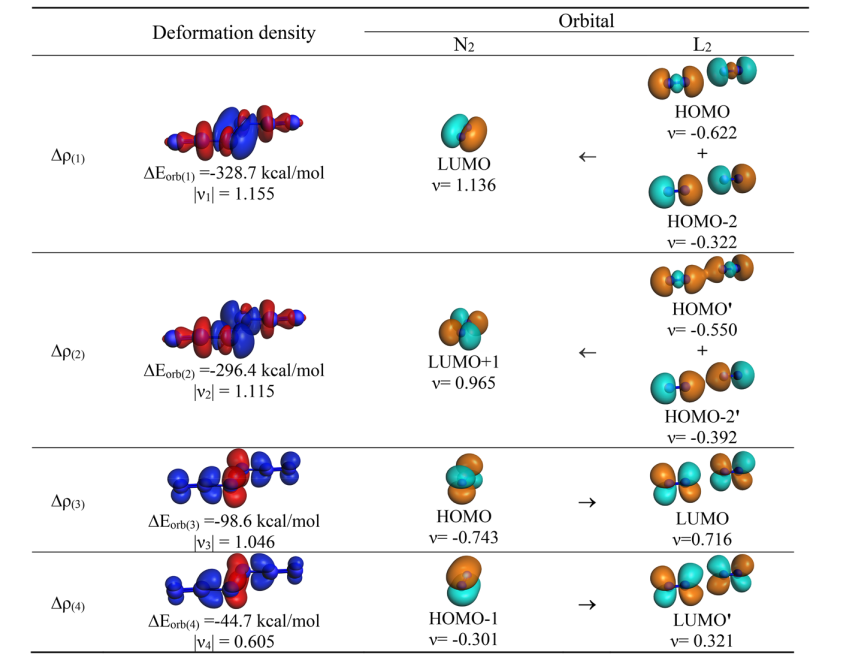
对于其他配体,最优片段组合存在差异:L = CO、CS、NO⁺ 表现为共用电子对双键(electron-sharing double bonds);对于 L = CN⁻,最低 △Eorb 值出现在处于电子四重态(4Σu+)的 N2 与作为对称性匹配四重态的末端 (CN)22⁻ 配体之间(表 3)。为横向对比,研究统一采用“激发态中心 N₂+基态配体”模型,其总相互作用能趋势(CS > CO > CN⁻ > NO⁺ > N2)与反应能垒趋势完全一致,证实了该模型的有效性。
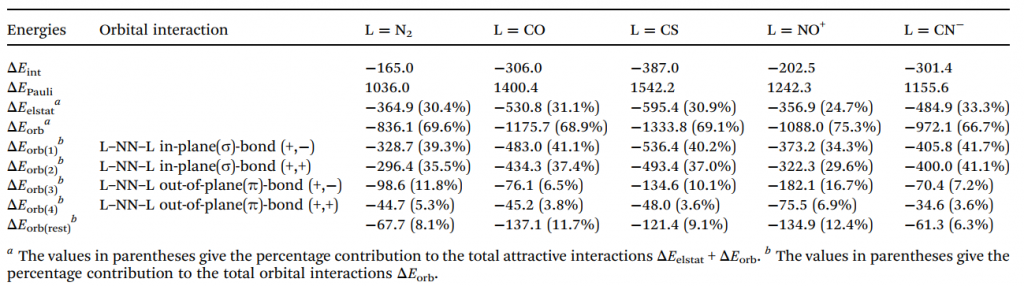
在此类中性体系的轨道作用中,σ 给予始终占据绝对主导地位。这导致了一个反常现象:CO 的 π 反馈贡献甚至低于 N2,背离了传统过渡金属配合物中的认知,其根本原因在于超强的 σ 给予作用掩盖了 π 反馈的差异。此外,静电作用、Pauli 排斥以及因中心键长变化引起的片段轨道弛豫,共同决定了分子的最终稳定性和键强趋势。
总结与展望
本研究在 CCSD(T)/cc-pVTZ 高精度水平下系统解析了 N2L2 (L = N2, CO, CS, NO⁺, CN⁻) 系列分子的结构与成键机制,确证所有分子的唯一构型均为反式共面排列。研究突破了传统基于 Lewis 结构式的定域双键认知,电荷分布与 EDA-NOCV 分析证实此类体系的核心驱动力为配体到中心 N2 的 σ 电子给予作用,而
π 反馈极弱。基于这种给体-受体配位模型,研究合理阐明了 N6 等分子以最低能垒协同解离为三个自由 N2 分子的实验现象。该工作不仅澄清了此类特殊配合物的电子结构争议,更为未来在实验上设计、合成及捕捉高活性N2活化产物奠定了坚实的理论框架。
参考文献
- Li Y, Ding C, Xie L, et al. Dinitrogen complexes N 2 L 2 (L= N 2, CO, CS, NO+, CN−)[J]. Chemical Science, 2026. DOI: 10.1039/d5sc08399k
感谢胡钧员老师供稿!