
背景
过去二十年,金(I)络合物因其优异的 π 活化能力,在均相催化领域大放异彩。特别是在炔烃的活化转化方面,阳离子型金(I)络合物已被广泛应用。然而,中性、配位饱和的金(I)络合物通常被认为催化能力弱、难以实现插入反应等过渡金属常规反应步骤。尽管已有零星研究报道了金(I)–X 键(X 为氢或杂原子)与炔烃发生插入反应,但几乎全部集中在单金属体系,且普遍需要高温或过量底物。近日,西班牙塞维利亚大学 Pablo Ríos 教授团队联合马德里康普顿斯大学 Israel Fernandez 教授在 Chemical Science 期刊上发表最新研究,首次系统性地揭示了双金(I)炔基络合物在插入反应中的独特反应路径,并通过实验和理论结合,阐明了第二个金原子如何降低能垒、改变反应方式。这项研究不仅刷新了人们对金(I)络合物反应性的认知,也为构建多金属协同催化体系提供了新思路。
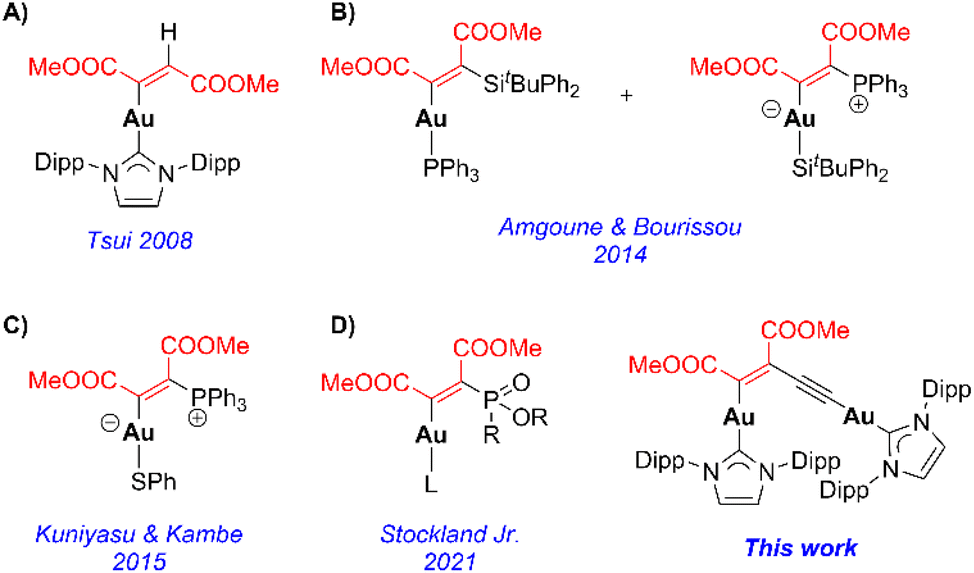
研究内容
研究者合成了一类以 NHC(IPr 与 SIPr)为配体的双金(I)炔基络合物(化合物 7 与 8),并通过 X 射线单晶衍射确认其结构,合成方法如图2所示。这些对称型络合物在室温下即可与电子缺陷炔烃 DMAD 反应,生成 Z-构型烯炔产物。而对比实验发现,单金属络合物(如IPrAu–C≡CSiMe₃)在相同底物下需要 110°C 以上高温才可实现插入。不仅如此,研究团队对反应动力学进行了系统分析:双金体系在 25°C 下即可完成反应,表观活化自由能 ΔG‡ 仅为 22.6 kcal/mol;SIPr 取代后的 8 号络合物因电子密度提高,反而反应稍慢,这表明金–配体反作用也起关键作用;单金体系反应活化能高达 30 kcal/mol,难以在常温下进行。
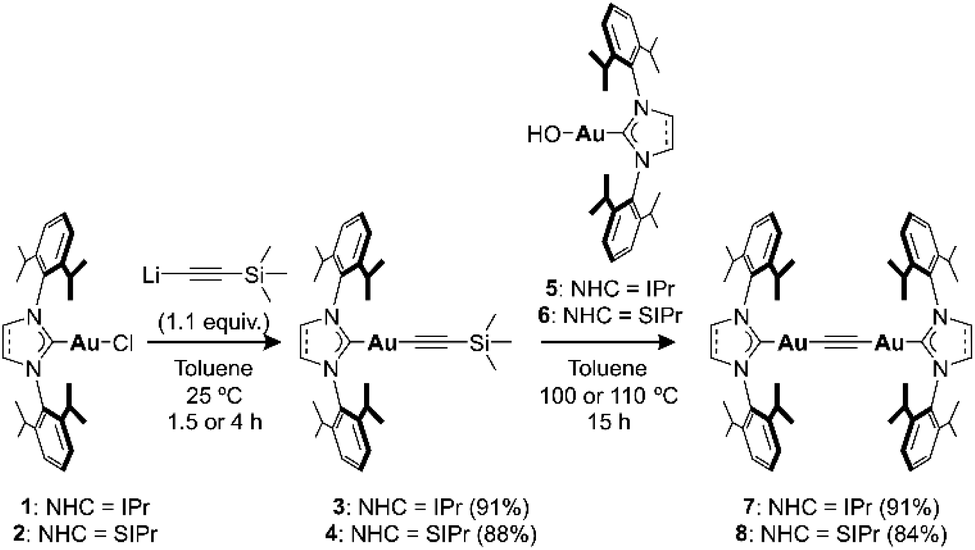
理论计算则揭示了一个前所未有的“分步插入机制”,而非文献中常见的协同插入路径:DMAD 的 LUMO 为 C≡C 键的 π* 反键轨道,而 7 的 HOMO 主要分布在炔基 C≡C 与两个邻近金原子上,表现出显著的 π 电子密度。因此,在反应初始,DMAD 以典型亲电方式进攻富电子炔基片段,通过一个关键过渡态(TS1)形成新的 C–C σ 键,该过渡态的能垒约为 19.5 kcal/mol,和实验测定的 22.6 kcal/mol 高度吻合,证实这是反应的速率决定步骤;第二步生成带有两个金原子的 s/p 混合中间体(σ,π-digold 结构);第三步经过构象转化与金原子迁移,最终生成 Z-构型烯炔插入产物。这一机制中,第二个金原子不仅增强了炔基碳的亲核性,还稳定了中间体,降低了关键过渡态的能垒。
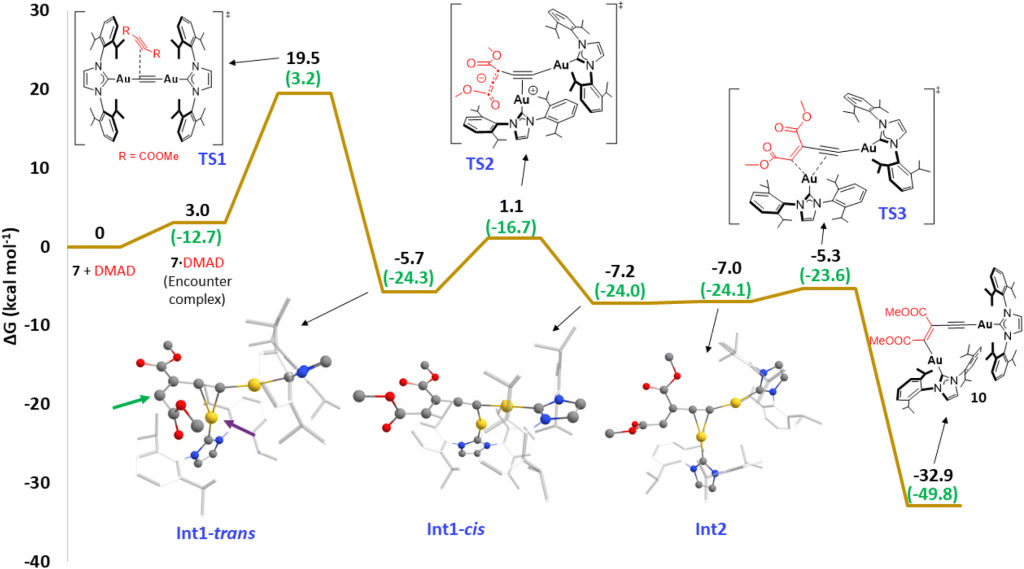
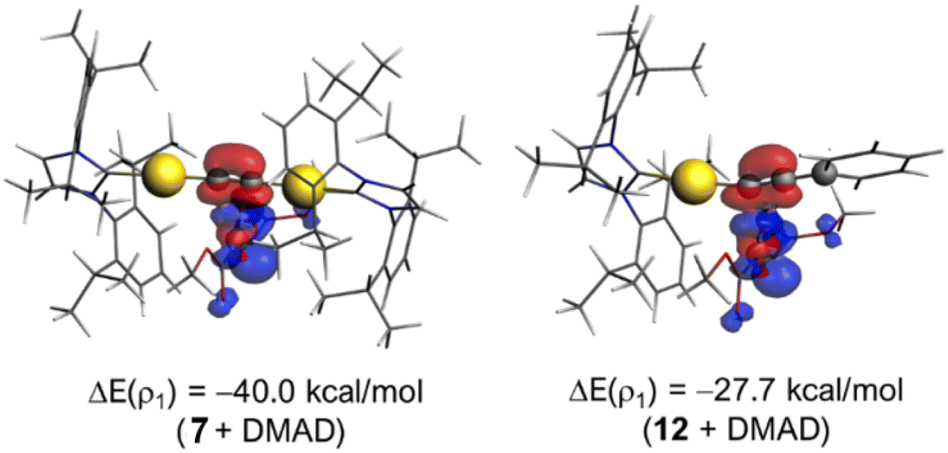
进一步的活化应变模型(ASM)与基于 AMS 软件的 EDA–NOCV 分析表明,双金络合物在 C–C 键形成的关键过渡态中,双金系统的静电相互作用(ΔE_electrostatic)与轨道相互作用(ΔE_orbital)显著更强,因电子极化效应,表现出更强的静电作用与轨道重叠,尤其是 π(HOMO) → π*(LUMO) 的作用大幅增强,是反应加速的核心。
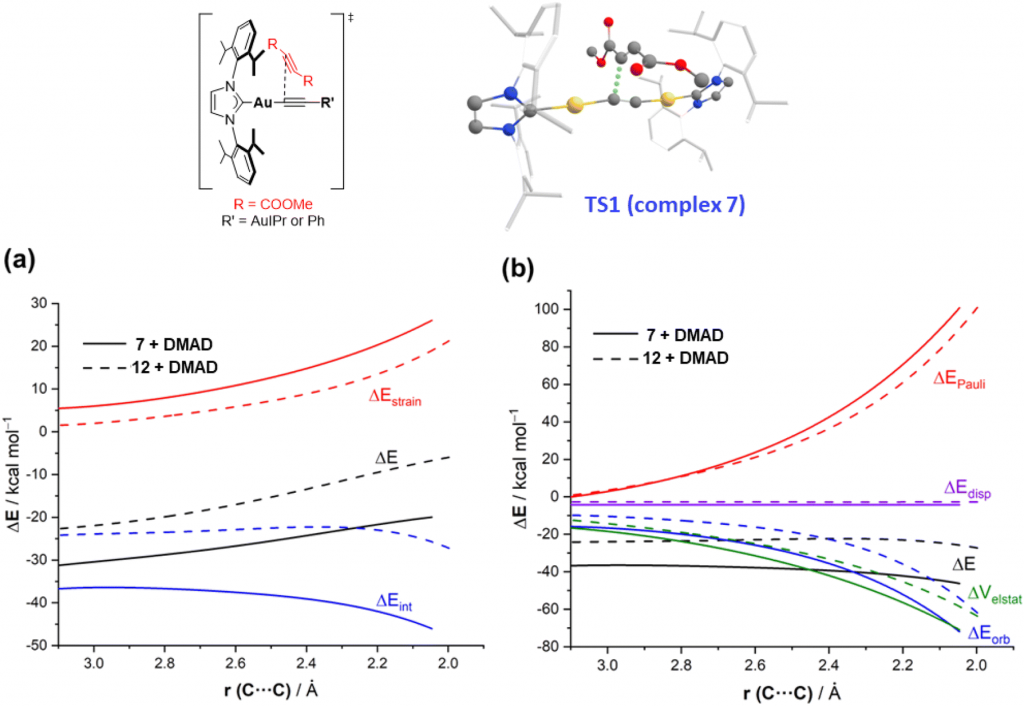
图 4.(上图)各机理中速率控制步骤的过渡态示意图,(下图)DMAD 与7(实线)和12(虚线)反应沿反应坐标投影到 C⋯C 成键距离的活化应变分析 (a) 和能量分解分析 (b)
结论
本研究突破性地构建了一类具有高反应活性的双金(I)炔基络合物,并首次揭示了其与炔烃底物发生插入反应的“分步机制”,不同于已有金催化反应中常见的协同路径。研究亮点包括:插入反应可在室温完成,远低于单金属体系的反应门槛; 第二金原子在电子赋能与中间体稳定中起决定性作用;DFT 计算结合能量分解分析精确揭示反应过程;所得机制可为多金属协同催化设计提供范式参考。这项工作不仅为基础金催化反应机制提供了新的认知视角,更拓展了构筑高效、多金属催化剂的可能性。未来,研究团队也计划在此基础上开发非对称双金体系或杂金属体系,用于更复杂的有机合成转化。
参考文献
- Cayuela-Castillo J, Fernández-de-Córdova F J, See M S, et al. Stepwise alkyne insertion in Au (i) acetylides: influence of the nuclearity[J]. Chemical Science, 2025, 16(11): 4684-4694. DOI: 10.1039/D4SC08227C
撰稿人:胡钧员老师