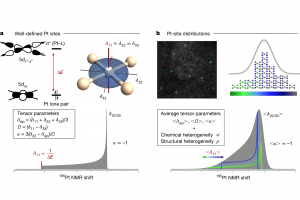
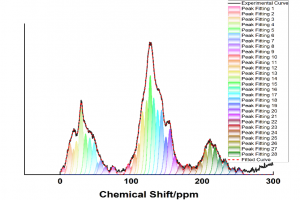
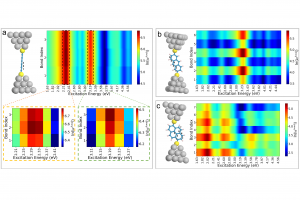
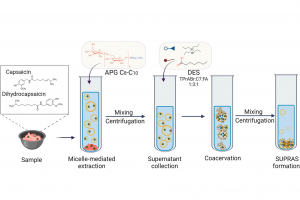
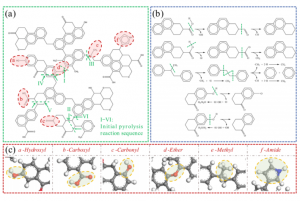
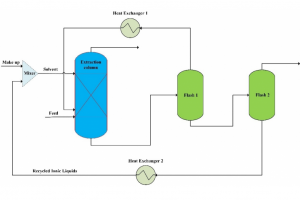
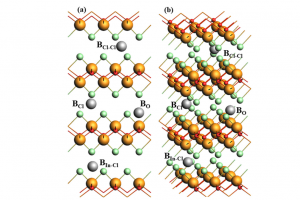
摘要 将原子分散物种与受控结构结合在一起的负载型金属催化剂,是催化材料设计的前沿领域,对反应性和金属高利用率提供了卓越的控制能力,接近分子级的精度。然而准确解析局部金属配位环境仍然面临挑战,它仍然阻碍着结构-活性关系认知的发展,而结构-活性关系是在不同应用领域中,优化设计所必需的信息。虽然电子显微镜能够揭示原子分散情况,但在多相催化中使用的传统光谱方法只能提供平均化的结构信息。里昂第一大学、苏黎世联邦理工学院、丹麦奥胡斯大学的研究者们,在最近发表于 Nature 的文章中,证明 195Pt 固态核磁共振(NMR)光谱是表征各种载体上原子分散 Pt 位点(即所谓的单原子催化剂 SAC)的有力工具。使用蒙特卡罗模拟,将 NMR 光谱转换为 SAC 特征信号,以分子精度描述配位环境,从而能够定量评估 Pt 位点分布情况及其均匀性。这种方法可以跟踪合成参数的影响,揭示特定步骤和载体类型的影响,还可以监测反应的变化,为具有目标结构的 SAC 可重复开发提供了关键见解。除了 SAC 之外,这种方法还为研究更复杂的结构奠定了基础,例如包含各种 NMR 活性金属的双原子或单簇催化剂。 本文使用 AMS2022 中的 ADF 模块对一系列 Pt(II) 模型配合物进行了几何结构优化,并计算了它们的 195 Pt NMR 光谱参数,计算所使用参数:PBE0 杂化泛函,中心 Pt 原子使用 QZ4P 基组,第一配位壳 Pt 使用TZP基组,其他原子使用 DZP 基组,相对论效应采用 ADF 中的 ZORA 方法处理。通过对参考化合物库的实验值和计算的各向同性化学屏蔽值进行线性回归,得到各向异性化学位移值。 其他相关研究 几乎同期,在美国的其他研究团队,使用 ADF 的相同的功能,在 J. Am. Chem. Soc. 也发表了非常类似的研究:99Ru Solid-State Nuclear […]