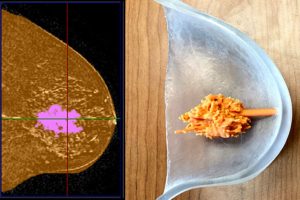
概述 3D打印解剖模型逐渐开始对医学产生重大影响。在本案例中,研究的对象是正在治疗肿瘤的乳房模型,因此患者能够更好地理解医生提出的诊断和后续治疗方案。同样地这些模型也有助于医生更好地观察的病理解剖结构。 亮点 ScanIP软件为肿瘤的可视化提供高效、精准的三维模型医生对3D打印模型的质量非常满意3D打印模型是理解治疗程序的重要工具模型为医生研究复杂手术的病理提供更多资源,从而减少手术时间和风险 介绍 癌症诊断始终都是一个难以面对的现实。对于大多数患者来说,面对陌生的医疗环境和医学术语,很难在没有“癌”的压力下进行思考。此外,大多数疾病的治疗高度依赖于患者对处方方案的依从性。这就需要患者和主治医生之间建立信任的伙伴关系,推进有效的沟通和可靠的随访。患者教育是建立准确认知所面临的挑战、治疗计划如何应对挑战以及患者和医生扮演不同角色的必要步骤。 3D打印的实物模型已被证明是一种高效的教育工具,能够更好地可视化病理解剖结构。本案例研究使用Simpleware软件中的分割工具快速生成一个乳腺癌患者的乳房和肿瘤的三维模型,由3D打印模型的展示通过患者教育建立共同基础。本案例还验证了增强医生对肿瘤位置和形状空间概念化的额外好处。 图:在Simpleware ScanIP中对MRI扫描的DICOM数据进行分割,生成2个3D模型:患者左侧乳房的模型和内部肿瘤的模型 MRI数据以DICOM格式获取并导入Simpleware ScanIP中进行分割。调整Window/Level设置实现最佳视图,使用Threshold工具设置灰度值范围,区别出广泛的乳腺组织和肿瘤。Crop工具可以将左侧乳房作为感兴趣区域单独裁切出来。分别在肿瘤掩模和乳房掩模上应用Mask Flood Fill工具,确保肿瘤和乳腺组织的分割清晰。 此填充的步骤确保掩模数据中只保留连通的体素。使用形态学的Close和Cavity Fill工具有效地填补掩模连续性上的空白,进一步优化掩模的完全覆盖。Paint工具允许用户通过主观认知判断对掩模进行最后的润色,确保按照需求对体素进行掩膜或去掉掩膜。在图像中完成乳房和肿瘤区域的遮挡,使用Resample工具对体素进行各向同性渲染。先用Dilate使掩模平滑,然后用Recursive Gaussian Smoothing生成一个干净、平滑的面。最后将掩模导出为STL模型,以便在打印前做进一步处理。 模型生成 分割后导出两个stl面模型:乳房和肿瘤,然后将它们导入Autodesk公司的Fusion 360®中做打印前的进一步修改。将乳房模型设计为中空的,打印的部分将呈现普通的外形但没有内部填充,因此肿瘤模型可以单独展示也可以嵌套在乳房模型内部。打印完成后还设计了一个柱状物连接肿瘤和乳房模型。该柱状物能够保证打印的肿瘤准确地显示在正确位置,乳房模型被分为两部分,方便观察。 图:将乳房的3D模型处理为中空的,并设计一个柱状物在打印后将肿瘤模型固定到乳房模型上 3D打印 将模型导入Stratasys GrabCAD ™中进行排列、处理,然后发送到3D打印机。这些模型是在Stratasys J750™打印机上使用Vero材料打印的,可以是选择从透明到黑色的多种颜色。 图:乳房模型用透明材料打印,肿瘤则是不透明材料。柱状物可以使肿瘤模型快速、准确地固定在乳房模型内。这些模型易于操作,可作为视觉辅助工具呈现。 模型的三个部分很容易组合在一起,乳房的两部分也能够对齐。在之后的迭代中还可以添加设计特征,如果有需要也可以把它们结合起来。将带有柱状物的肿瘤模型安装在有设计预留柱孔的乳房模型,使得模型间的配准简单又快捷。将柱状物放在孔洞后,肿瘤即可处于相对乳房模型的适当位置。乳房模型中肿瘤的大小和位置是打印模型最重要的意义,因此一个包含肿瘤和乳房的比例模型和简单的空间定位机制就可以使设计即刻变得实用。 医生反馈 合作医生对打印结果印象深刻,并发现了超出本项目范围的益处。他们证实模型在协助外科医生规划和沟通切除途径和其他治疗方式方面的效用,加深患者对病理学含义理解的潜力也激发了医生。通过可视化增强患者的理解有助于医患之间就各种治疗方案的需求和影响进行对话。 例如,可视化肿瘤的大小和位置可以促进关于肿块切除术(切除肿瘤)或乳房切除术(切除整个乳房)决定的沟通。这种对话很敏感,而可视化则是一个有效的工具。此外,视觉辅助工具可以通过更好地了解治疗方案的目标来帮助提高患者的依从性和信心。 结论 这种可视化人体解剖学的方法具有深远的意义。在这里,我们讨论了对患者教育的影响,但在医生指导方面还有更多工作可以开展。同时也可以改进医生教育,因为病理模型和周围组织可以采用能够模仿其外观和触觉特性的材料打印。使用多模型、多材料打印制作患者个性化手术模型,将加强医生治疗复杂问题的能力,可能减少手术时间,减少对周围组织的附带损伤,通过直接在患者病理学模型上实践从而增强治疗效果。 高效地生成分割模型的另一个潜在用途是患者的个性化生物打印。对健康组织和病变组织进行建模获得的数据可用于生物打印替代组织,在之后植入以取代或再生感染区域。不同于将模型切片后用塑料打印,生物打印过程采用生物降解塑料和水凝胶,按照设计的组织模式打印多种细胞类型,生成特定的功能性组织。 3D打印解剖模型正逐渐开始对医学产生重大影响。患者个性化模型打印的第一步是患者影像数据集的分割,可打印模型由图像数据的分割部分快速产生,Simpleware ScanIP可以使该过程简单高效。包含治疗中肿瘤的乳房模型能够更好地理解医生提出的诊断和后续治疗方案,同时也有助于医生对病理解剖进行可视化。 参考 Kengla, C., Renteria, E., Wivell, C., Atala, A., Yoo, J.J., Lee, S.J., 2017. Clinically Relevant Bioprinting Workflow and Imaging […]