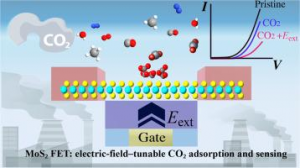
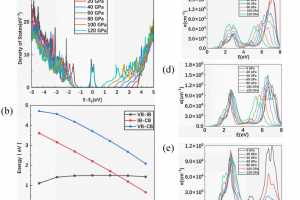
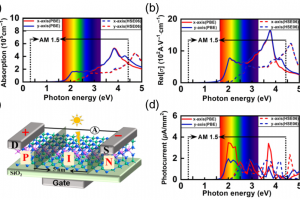
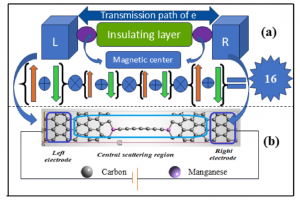
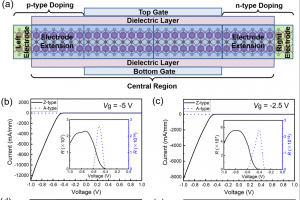
基于 WTe2 单层的超灵敏、可回收 FET 型有毒气体传感器 场效应晶体管(FET)型气体传感器因其功耗低、灵敏度高而吸引了大量研究人员的关注。然而,对其传感能力和内在机理的理论探索仍然十分匮乏。本文以单层纯 WTe2 和缺陷 WTe2 为传感平台,利用第一性原理计算和统计热力学模型,系统地研究了 FET 型气体传感器对 SO2、CO、NO、NH3 和 NO2 等多种有害气体的传感特性和工作原理。研究结果表明,基于纯 WTe2 的 FET 型气体传感器在零栅压下工作时,对 NO2 的检测具有极高的灵敏度和重复使用性,在 20 ppb 低浓度下的灵敏度达到 96%。引入 Te 空位是一种高效的策略,可以提高气体传感器对所有测试有毒气体的灵敏度,同时确保其可重复使用性。在基于纯 WTe2 的 FET 型气体传感器上施加栅压可进一步提高其对 NO2 的灵敏度,在整个偏压范围内超过 90%,在 3 V 栅压下达到 97%。本研究为实现 ppb 级浓度有毒气体的超灵敏和可回收检测提供了一种前景广阔的方法。 Journal of Materials Chemistry A, 2024, 12(39): 26951-26961. DOI :10.1039/D4TA02739F 金修饰的黑磷:氮氧化物去除剂和氢传感器 有效检测有毒气体和清洁能源氢气是工业生产和日常生活的迫切需要。本文通过密度泛函理论结合非平衡格林函数方法,从理论上研究吸附/掺杂金的黑磷单层对六种不同气体(CO、H2、H2S、NO、NO2 和 SO2)的传感特性。计算得出的吸附能、电荷转移、电子局域函数、能带结构和态密度表明,吸附/掺杂金的黑磷对氮氧化物气体表现出敏感的化学吸附。而令人惊讶的是,掺杂金的黑磷对 H2 分子表现出敏感的物理吸附。计算得出的恢复时间和电流电压曲线表明,吸附/掺杂金的黑磷器件是一种超灵敏(灵敏度为 71%-100% )的氮氧化物分子去除器。更重要的是,掺杂金的黑磷传感器可在室温下实现对 H2 的高灵敏度(灵敏度为 85%)、选择性和可重复使用性(恢复时间为 0.01 ns)检测。本研究结果为吸附/掺杂金的黑磷在气体传感领域的潜在应用提供了理论依据,尤其是在理想的氮氧化物去除剂和 H2 传感器方面。 Applied Surface Science, 2024, 651: Article 159194. DOI:10.1016/j.apsusc.2023.159194. 利用新型二维五边形 Pd2P2SeX(X= O、S、Te)pin 结纳米器件设计高性能场效应、应变/气体传感器:传输特性研究 在现代纳米器件领域,获得高开关性能并在高精度环境中实现传感检测功能至关重要。二维(2D)Pd2P2SeX(X = O、S、Te)具有优异的几何、物理和化学特性,可以结晶成具有间接带隙的正交结构。基于这些基本特性,通过第一性原理对二维 Pd2P2SeX pin 结器件进行建模以模拟场效应晶体管、高开关比应变传感器和基于化学吸附的气体传感器。Pd2P2SeX pin […]