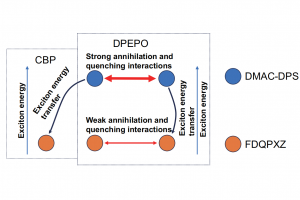
研究背景 过去几十年间,有机发光二极管(OLED)因其广色域、超高对比度、低能耗和柔性特性,已在显示市场获得广泛应用。高效白光有机发光二极管(WOLED)作为大尺寸照明光源、液晶背光和标识应用的潜力正日益凸显。尽管 WOLED 具备显著的应用前景,要实现商业化仍需攻克多项技术难题,其中提升发光效率尤为关键。要实现多层堆叠结构 WOLED 的理性设计与优化,必须对器件物理机制(尤其是电荷与激子动力学)建立精确的定量认知。构建完整 OLED 结构的物理模型面临以下几大挑 战: 真空或溶液工艺制备的分子薄膜具有非晶态特征; 发光层及电荷传输层所采 用的分子混合材料; 激子过程中复杂的物理机制,涵盖激子的生成、传输及相互作用; 覆盖从纳秒级光物理过程到亚毫秒级载流子传输以及低迁移率层状材料中弛豫过程的广泛时间尺度 虽然宏观连续体漂移-扩散模型可以在器件尺度上描述电流密度, 但在应对主客体分子的分子级混合及其对激子过程速率的影响时仍存在局限。而基于密度泛函理论的微观原子级模型能够揭示单个分子或晶体系统的电学与光学特性,但将此模型应用于完整 OLED 堆叠结构的仿真仍充满挑战。 研究内容 华南师范大学华南先进光电子研究院彩色动态电子纸研究所刘飞龙课题组围绕 2019 年华南理工大学在Nature Communications 报道的一种高性能 TADF 型白光 OLED 器件展开研究。该文章中提出了一种高效发光的荧光 WOLED 器件,并且做了三种不同的器件来证明器件的高效性。该器件在合理的结构设计下实现了约 20.5%的外量子效率和优异的器件寿命,展现出良好的应用前景。然而,该文章并未对其中的橙色 TADF 发光材料 FDQPXZ 是如何敏化蓝色 TADF 材料 DMAC-DPS 这一关键过程进行深入探讨。理解这一能量转移过程的底层物理机制,对于未来设计更高效率的 WOLED 器件至关重要。 本研究通过 Bumblebee 软件进行三维动力学蒙特卡洛模拟,仅使用一组材料参数就成功复现了多种实验表征良好的 TADF WOLED 堆叠结构的性质,包括:电流密度-电压特性、外量子效率滚降特性及瞬态光致发光特性。针对具有混合发光层和多种器件设计的复杂多层 OLED 堆叠结构同步优化提取了电子和激子器件参数。该研究定量揭示了电子能级结构、激子能量与电荷传输及激子过程速率之间的复杂相互作用机制,系统阐释了所研究器件的高效机理。基于这些物理认知进一步提出了具有更高效率的创新器件设计方案。 图 1 器件 1 优化前后能级示意和空穴分布图 2019 […]