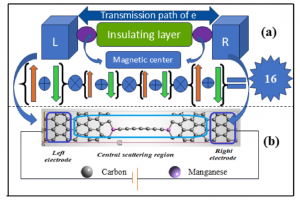
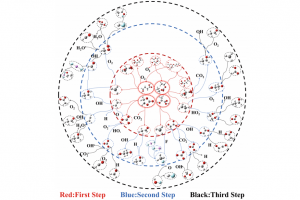
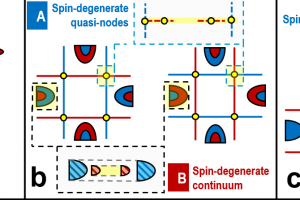
研究背景 随着信息技术的发展,数据存储器件正朝着更高速度、更低功耗以及更高稳定性的方向演进。磁阻随机存储器(MRAM)由于能够同时满足这些需求,被认为是最有潜力的下一代非易失存储技术之一。MRAM的核心器件是磁隧穿结(magnetic tunnel junction, MTJ),其基本结构由两层磁性电极和中间的绝缘势垒层组成。当两侧电极的磁矩由平行变为反平行排列时,电子的隧穿概率会发生变化,从而导致器件电阻出现明显差异。这种由磁构型改变引起的电阻变化被称为隧穿磁阻(tunneling magnetoresistance, TMR),是评价 MTJ 性能的重要指标。 传统 MTJ 器件通常采用铁磁材料作为电极。然而,铁磁体具有宏观净磁矩,会产生杂散磁场,这在器件小型化和高密度集成过程中可能带来干扰,影响器件稳定性。另一方面,反铁磁材料虽然没有杂散磁场,但其电子结构通常缺乏明显的自旋极化特征,使得自旋输运能力较弱。近年来提出的交错磁(altermagnetism)为这一问题提供了新的解决思路。交错磁材料在磁结构上类似反铁磁体,没有宏观净磁矩,但其能带结构却呈现出类似铁磁体的自旋分裂特征,从而兼具两者的优势,为新型自旋电子器件的设计提供了新的可能。 研究内容 在磁隧穿结中,电子通过势垒层进行量子隧穿时需要同时满足自旋守恒和动量守恒,因此电极材料在费米能级处的自旋分辨费米面几何直接决定了平行(P)与反平行(AP)构型下的导电通道。如图 1所示,当两侧电极的导电通道在动量空间发生重叠时,AP 态仍存在可用的自旋简并通道,从而产生“漏电流”,并限制隧穿磁阻(TMR)的提升。理想情况下,若两种自旋的费米面能够完全分离,则反平行态的输运将被极大抑制,从而获得更高的磁阻效应。 基于这一物理机制,北京大学物理学院吕劲团队与新加坡科技设计大学 Yee Sin Ang 研究组合作,系统研究了交错磁材料中费米面结构对交错磁隧穿结(AMTJ)输运性质的影响。研究选取了三种已经在实验中成功合成的交错磁材料:V2Te2O、RbV2Te2O 和 KV2Se2O,并通过密度泛函理论结合第一性原理量子输运计算,对其器件输运性质进行了系统模拟。 研究发现,准层状材料 RbV2Te2O 和 KV2Se2O 在费米能级附近存在交错磁平带结构,这种平带会产生高度各向异性的准二维费米面。如图 2 所示,随着材料原子构成的变化,不同自旋导电通道在动量空间中的重叠区域逐渐减少。在 V2Te2O 中 为 Γ 点附近的延伸区域,在 RbV2Te2O 中缩小为弧形区域,而在 KV2Se2O 中则仅剩少数离散节点。后两者材料的这种费米面几何结构能够显著减少自旋通道的重叠,因此可以有效抑制反平行态下的隧穿电流,从而大幅提升器件的磁阻效应。 图 1:不同电极费米面形状对交错磁隧穿结输运性质的影响 图 2:(a)交错磁隧穿结(AMTJ)在平行(P)与反平行(AP)构型下的磁矩排列示意图;(b-d)分别为基于 V2Te2O、RbV2Te2O 和 KV2Se2O 电极构建的 AMTJ 器件在费米能级处的自旋极化 k 分辨透射系数分布 在三种候选材料中,KV2Se2O 表现出最理想的费米面自旋分离特征。费米能级附近的不同自旋输运通道几乎互不重叠,为实现高磁阻提供了关键条件。基于这一性质,研究团队以 KV2Se2O 为电极构建 AMTJ 器件,并系统评估其输运性能。模拟结果显示,即使在简单的真空势垒结构下,器件的 TMR 已达到约 4.3 × 103 %。当进一步引入与电极晶格和对称性匹配的绝缘势垒后,TMR 可提升至 1.1 × 106 %,显著超越了传统铁磁 MTJ 以及其他代表性器件的性能(图3)。 图 3:(a)基于 KV2Se2O 电极的 AMTJ 器件性能与其他代表性 MTJ 的对比(b)上述不同器件对应的结构设计与研究方法(c)典型 MTJ 器件结构示意图 总结 本工作从费米面几何结构出发,系统阐明了交错磁平带在抑制动量空间自旋通道交叠、提升隧穿磁阻方面的核心作用。其中,以 KV2Se2O 为电极的交错磁隧穿结在对称匹配的绝缘势垒辅助下,可以实现高达 1.1 × 106 %的理论 TMR,显著突破现有 MTJ 体系的性能上限。本研究不仅确立了交错磁平带驱动的费米面工程设计范式,也为新一代高密度、低功耗自旋存储与自旋逻辑器件的发展提供了重要的材料与结构指导。 参考文献 X. Yang, S. Fang, Z. Yang, P. Ho, J. Lu, and Y. S. Ang, Altermagnetic Flatband-Driven Fermi Surface Geometry for Giant Tunneling Magnetoresistance, Adv. Funct. Mater., e31921 (2026). http://doi.org/10.1002/adfm.202531921