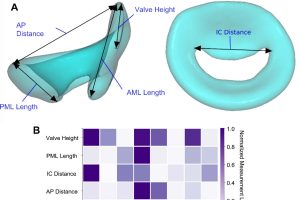
概述 二尖瓣反流(MR)影响着全球超过 2400 万人,是发病和死亡的主要原因之一。若不及时治疗,可能会进展为心房颤动、肺动脉高压,最终导致心力衰竭。经导管二尖瓣置换术(TMVR)因二尖瓣解剖结构的复杂性和高度变异性而面临独特挑战。为确保手术成功,TMVR 器械必须实现安全锚定、有效封堵和左心室流出通畅。然而,目前的临床前设计范式因对人群水平的二尖瓣解剖变异性认识不足而受到阻碍。 统计形状模型(SSM)为量化解剖结构变异性和构建具有代表性的 3D 解剖虚拟患者队列提供了一个稳健的框架,可作为生物力学计算机模拟临床试验的基础。本研究利用 72 例成人的前瞻性心电门控对比增强冠状动脉 CT 血管成像(CCTA)扫描数据开发了一个二尖瓣的统计形状模型,主成分分析揭示了形状变异的主要模式,与文献中已报道的解剖模式一致,验证了模型的生理相关性。SSM 能够有效捕捉二尖瓣的几何多样性,无需对标志点或线性测量的重要性做出任何预设,结果证明了 SSM 由现有扫描数据生成虚拟患者群体的实用性。这些发现支持将 SSM 整合到用于临床前测试、器械设计和个性化医疗的建模流程中。 图像处理 选取 orCaScore 数据集中 72 名成人(男女数量相等)的 CCTA 扫描数据,采用心电图触发将扫描采集同步到舒张静止期,具体为 R–R 间期的 70%(GE、西门子)、75%(东芝)或 78%(飞利浦)。值得注意的是,本统计形状模型仅反映受试者之间的解剖变异性,并不包含周期内(时间)变形。所有患者均无解剖异常、冠状动脉内支架和金属植入物,且扫描图像无严重运动伪影和高噪声水平。 使用 Simpleware 软件的心脏自动分割工具 AS Cardio 对原始 CCTA 数据进行处理,一键点击获得主要的心脏腔室。然后应用软件中的自定义后处理脚本从中分割出二尖瓣环、前叶、后叶。所有分割结果均经过人工审查,并使用 Simpleware 软件的区域生长和网格编辑工具对微小缺陷如孔洞、分割噪声或边缘伪影进行校正。 图:从 CCTA 分割到 SSM 的工作流程概述 统计形状建模 为限制扫描采集中旋转和平移变化的影响,在每个分割上手动放置四个解剖标志点应用配准算法。沿二尖瓣环的前后轴定义两个标志点:(1)二尖瓣前叶(A2 扇叶)中点之上的瓣环最上点(2)二尖瓣后叶(P2 扇叶)中点之上的瓣环最上点。其余两个标志点为与前内侧连合(A1–P1 交界处)和后外侧连合(A3–P3 交界处)对应的瓣环铰链点。将这些标志点用作最佳拟合刚性变换的点,通过迭代最小化对应点之间的距离对齐两个表面,有效消除平移和旋转差异的同时保留比例和几何形状。对对齐后的分割结果进行主成分分析(PCA),形状变异性被评估为相对于参考几何形状的顶点几何变化。 为表征样本群体中的解剖变异性,评估二尖瓣的关键几何尺寸作为沿形状模式变化的函数。测量二尖瓣前叶(AML)长度、二尖瓣后叶(PML)长度、瓣环前后径(A–P)、连合间距离(IC)和瓣膜高度(二尖瓣前叶最上点与前内侧和后外侧连合所确定平面之间的垂直距离)。使用 Simpleware 软件的测量工具和 SSM 工具包将测量值从参考模型自动映射到每个输入模型。为便于自动测量映射,从参考图像中提取代表上述测量的 […]