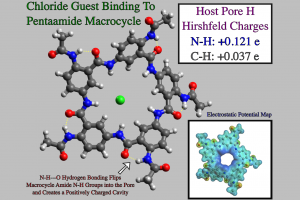
如果主体对携带特定电荷的客体物种具有亲和力,则可能用于对于对抗各种疾病。合成的超分子结构被设计为与阳离子或阴离子结合,利用官能团的影响得到大环的孔,以获得负电荷与阳离子相互作用,或正电荷以结合、运输阴离子。通过迫使孔隙带正电,让生物膜主体具有阴离子结合特异性,这对于对抗氯离子输送故障和治疗囊性纤维化非常重要。在最近的一项研究中,主体五酰胺大环被特异官能化,通过N-H–O氢键迫使酰胺基团被定向到孔中,如此将相当大的正电荷放入五酰胺的空腔中,并将羰基翻转到大环之外。由于孔中 NH 和 CH 的正电荷,这些五酰胺大环对阴离子具有特异亲和力。 五酰胺大环上的 Hirschfeld 电荷和静电势,以及单体构建块的偶极矩,使用 ADF 计算得到,证明了腔的强正电性。几何优化表明,过量阴离子客体被吸引到五酰胺大环的正电空腔中。计算的缔合常数和结合能表明,主体空腔偏向去溶剂化,以允许孔内容纳阴离子客体。本文特别关注了主体结构内氯化物、溴化物和碘化物客体的结构优化,计算表明碘化物与五酰胺腔的结合最强,其次是氯化物,溴化物的结合最弱。这与 N-H–Cl– 相互作用强于 N-H–Br– ,N-H–I– 相互作用最弱的一般趋势相反。结构优化表明,碘化物与五酰胺大环的结合最强,因为它的尺寸更大,能够与孔中的所有N-H基团相互作用。由于氯化物和溴化物的尺寸较小,这两种阴离子只能聚集两个 N-H 基团配位。考虑到氯化物比溴化物的结合力强,又不像碘化物那样过度填充孔隙,因此相对于其他卤化物,氯化物的传输能力最好。 因此,正如ADF计算结果所显示的那样,五酰胺大环是对抗囊性纤维化和补充氯化物正常运输的理想选择。 参考文献 Ruikai Cao, Robert B. Rossdeutcher, Yulong Zhong, Yi Shen, Daniel P. Miller, Thomas A. Sobiech, Xiangxiang Wu, Laura Sánchez Buitrago, Karishma Ramcharam, Mark I. Gutay, Miriam Frankenthal Figueira, Pia Luthra, Eva Zurek, Thomas Szyperski, Brian Button, […]