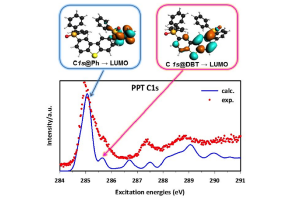
单通道的Kohn-Sham DFT理论结合跃迁势(Transition Potential),能够考虑核空穴形成的大部分的电子弛豫效应,从而描述轻原子的K-壳层NEXAFS光谱。这种方法提供了一组正交轨道,从中可以得到跃迁偶极矩。 K壳层NEXAFS光谱是通过对每个非等效原子位的激发光谱进行单独计算,并将其贡献按相对权重相加得到的。这样就可以将总的光谱性质反卷积到不同组分中,从而有助于将光谱特征分解到分子的特定部位。 这种方法,最近被应用到2,8-bis-(diphenylphosphoryl)-dibenzo[b,d]thiophene(PPT)的C1s和O1s NEXAFS光谱的模拟,这是最近引入OLED中的一种双极性磷光主体材料,用于解释在Trieste的电子同步加速器气相束线处获得的实验光谱。PPT可以认为是由两个二苯基氧化膦(dPPO)部分,对小二苯并噻吩(DBT)核心官能化而形成。 在C的K-边的DFT-TP计算表明,PPT的C1s谱主峰归属于dPPO臂的苯环,而第二弱峰则归属于PPT DBT核的苯环部分。 本研究的结论对OLED的未来应用具有重要意义:PPT的氧化膦基团是PPT的DBT核与外层基团之间π共轭的断裂点。然而,这些基团在很大程度上不影响DBT中心部分的电子性质。 参考文献: A. Guarnaccio, T. Zhang, C. Grazioli, F. Johansson, M. Coreno, M. de Simone, G. Fronzoni, D. Toffoli, E. Bernes, C. Puglia, PPT Isolated Molecule and Its Building Block Moieties Studied by C 1s and O 1s Gas Phase X-ray Photoelectron and Photoabsorption Spectroscopies, J. Phys. Chem. C […]