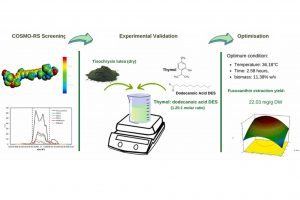
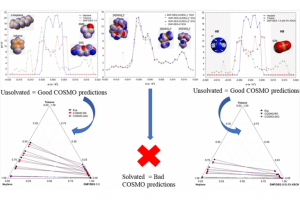
研究亮点 使用COSMO-RS筛选用于提取岩藻黄质的24中共晶溶剂通过实验验证了COSMO-RS筛选得到的6个排名靠前的共晶溶剂百里酚 : 十二酸共晶溶剂的岩藻黄质提取率最高利用σ-profile和σ-potential揭示了可能的岩藻黄质提取机制最佳条件下岩藻黄质提取率为 22.03 mg/g DW 研究内容 岩藻黄质是藻类中的一种类胡萝卜素,据称具有抗氧化、抗光老化、抗转移、抗高血压活性等有益的健康相关特性,从而可能应用于化妆品、饮食和医药等。 奥克兰大学Dingmeng Xu等,研究评估了使用共晶溶剂 (deep eutectic solvents,DES) 从微藻Tisochrysis lutea中提取岩藻黄质的方法。 使用COSMO-RS筛选24种不同类型的共晶溶剂在提取岩藻黄质中的性能,然后使用6个排名靠前的共晶溶剂进行实验提取验证。 实验结果表明,其中百里酚 : 十二酸(摩尔比 1.25 : 1)对岩藻黄质的提取能力最高(7.69 mg/g dry biomass weight (DW)),高于常规溶剂甲醇 (6.29 mg/g DW) 和乙醇 (6.75 mg/g DW),这与 COSMO-RS 筛选结果一致。 作者进一步优化了百里酚 : 十二酸提取岩藻黄素的提取条件,结果表明在温度36.2 ℃、搅拌时间2.58小时,生物量百分比 11.36%时,岩藻黄质的提取率最高(22.03 mg/g DW)。 此外,岩藻黄质在百里酚 : 十二酸中表现出良好的稳定性,储存时间超过 11 天。经过七个提取周期后,最终岩藻黄质浓度 (13.06 mg/mL DES) ,可重复使用性良好。 参考文献 Evaluation of Deep […]