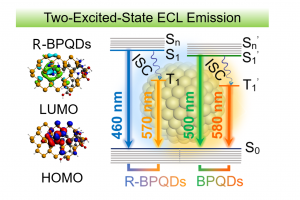
电化学发光(ECL)过程中的激发态是通过电活性物质与电极之间发生电子传递形成的中间体自由基之间发生电子转移反应产生的。通常,激发态物种可以是1R*或者3R*。根据自旋统计规则, 1R*和3R*的比值为1:3,从最低激发三重态(T1)到基态(S0)是非辐射跃迁。因此,ECL发光体的最大量子效率仅为25%,这限制了ECL过程中辐射能量的充分利用。热激活延迟荧光过程可以实现3R*到1R*的快速反系间窜越,从而充分利用单线态和三线态激子。但是,热激活延迟荧光过程通常仅在最低激发单重态(S1)和T1之间的能隙足够小(约0.1 eV)时才能发生。近期,南京大学生命分析化学国家重点实验室的鞠熀先教授研究团队用黑磷量子点(BPQDs)作为ECL发光体,实现了同时从S1到S0和T1到S0发生辐射跃迁产生ECL。 BPQDs在2014年首次由块状黑磷制备(Angew. Chem., Int. Ed. 2015, 54, 3653−3657),已广泛用于光热治疗、电催化和柔性器件等领域。BPQDs和黑磷纳米片的ECL已受关注,但空气中的氧化降解使黑磷纳米材料表面存在氧化缺陷,严重影响其光学和电学性能。在该工作中,作者用溶剂热法制得直径4 nm左右的BPQDs(图1),提出一种精氨酸(Arg)钝化BPQDs氧化缺陷的策略。精氨酸通过其胍基的离域正电荷与BPQDs表面氧化缺陷的负电荷间的静电和氢键相互作用结合,提高了BPQDs的水溶性、稳定性、可修饰性和ECL性能。 图 1. 精氨酸修饰前(BPQDs,蓝色)后(R-BPQDs,红色)的电镜与谱学表征 在阴极共反应剂K2S2O8存在时,R-BPQDs/GCE在−1.20 V出现ECL峰,其强度比BPQDs/GCE高25倍,阴极ECL效率比BPQDs高3.2倍。BPQDs和R-BPQDs的ECL光谱都表现出双发射的性质,其波长与它们各自的荧光和磷光峰位置一致。因此,BPQDs和R-BPQDs具有同时从S1到S0和T1到S0发生辐射跃迁产生ECL的性质(图2)。根据精氨酸与BPQDs的氧化缺陷之间的静电和氢键相互作用,作者用AMS软件ADF模块进行密度泛函理论计算,验证了精氨酸的重要作用。氧化缺陷导致BPQDs的最高占据分子轨道(HOMO)局域在缺陷位置,阻碍电子转移,因此降低了发光强度。在精氨酸修饰钝化表面氧化缺陷后,R-BPQDs的HOMO移动到中心位置,从而改变了电子跃迁途径,使HOMO与最低未占据分子轨道(LUMO)的空间重叠更大,明显提高了发射强度和电荷转移能力。通过比较不同氨基酸修饰BPQDs后的ECL变化,作者认为R-BPQDs的阴极ECL增强归因于精氨酸中吸电子胍基的存在,它稳定了邻近的电子注入R-BPQDs的LUMO后产生的R-BPQDs阴离子自由基R-BPQDs•−。 图2. 精氨酸修饰BPQDs前后的阴极ECL机制 为了将R-BPQDs的ECL用于生物分析,作者用含有精氨酸的多肽(精氨酸-精氨酸-甘氨酸-天冬氨酸-丝氨酸,RRGDS)代替精氨酸修饰BPQDs,合成RRGDS-BPQDs,其中RGDS可以特异性识别整合素αV/β3,并将RRGDS-BPQDs共价连接在多壁碳纳米管修饰的GCE上,利用RGDS将细胞膜表面富含整合素αV/β3的A549细胞结合到电极表面,开发出一种用于评估整合素抑制剂抑制效率的灵敏ECL方法,证明了BPQDs的ECL以及激发态调节策略的实用性。该工作提出的ECL激发态调节新策略为解密ECL机理提供了新途径,并拓宽了纳米ECL发光体的应用。 上述相关成果已以“Arginine-modified black phosphorus quantum dots with dual excited states for enhanced electrochemiluminescence in bioanalysis”为题发表于Nature Communications(DOI: 10.1038/s41467-022-35015-9)。博士生于思琦和博士后杜宇为该工作的共同第一作者,鞠熀先教授为通讯作者。 鞠熀先教授课题组长期从事纳米发光体的ECL及其生物传感应用研究:首次在水相体系研究QDs的ECL性能,并将其用于共反应剂的化学传感(Anal. Chem. 2004, 76, 6871–6876),制得第一支量子点ECL生物传感器(Chem. Commun. 2007, 404–406),构建了QDs的能量转移、电子转移、表面缺陷、自生共反应剂、共反应剂消耗等ECL新机制,发现了新的ECL共反应剂,建立了小分子、DNA、蛋白质和糖基的ECL检测方法,研制出低ECL电位的量子点。随后,该研究组设计合成了多种具有ECL性能的无机纳米颗粒,如g-C3N4纳米片、钴基MOF、电活性MOF、鲁米诺或Ru(bpy)32+负载的纳米复合材料、双稳定剂封端的Au纳米簇和铜掺杂的铽MOF等,提出新的ECL生物传感机制(Angew. Chem. Int. Ed. 2020, 59, 10446-10450; J. Am. Chem. Soc. 2021, 143, 3049-3053; Anal. Chem. 2021, 93, 14878)。近年来,鞠熀先教授通过合成噻咯-咔唑偶联的聚合物量子点(Pdots)和钌联吡啶掺杂的Pdots,提出了双ECL增强策略,发展了电子受体-电子供体-聚集发光基团偶联的高效ECL发光体和Pdots双分子内共振能量转移的ECL体系,构建了金属离子和多种疾病标志物高通量可视化成像检测方法,用合成的共反应剂内嵌高ECL效率的Pdots,实现了单个活细胞膜蛋白与单细胞分泌物的无试剂ECL成像检测(Angew. Chem. Int. Ed. 2021, 60, 197; Biosens. […]