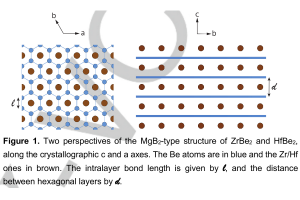
丹麦奥胡斯大学Maarten G. Goesten教授,近期发表了关于MBe2 (M=Zr, Hf)层状结构能带与层间相互作用能,及其超导性的研究结果。铍是一种s区元素,在ZrBe2和HfBe2合金中形成离域Be-Be π键的芳香族网络,从而构造出[Be2]4-层状堆叠,中间为+4价阳离子(如下图所示)。[Be2]4-亚晶格与石墨等电子、同构,MgB2中的[B]-2亚晶格也是如此,并且在其电子能带结构中与π键具有相同的表现。 它们在布里渊区的K和H处,出现能级的(近)简并,但由于层间轨道相互作用,而导致能量上的分离。Zr和Hf利用它们的价态d轨道,在层与层间形成键,导致几乎相同的能带结构。像MgB2一样,ZrBe2和HfBe2在环境压力下,经计算为声子介导的超导体,临界温度分别为11.4 K和8.8 K。声子和自由电子之间的耦合强度非常相似,因此临界温度的差异,由层间阳离子的质量(Mass)控制。 本文的DFT计算采用AMS软件BAND模块完成。 参考文献: Maarten Goesten, Be-Be π bonding and predicted superconductivity in MBe2 (M=Zr, Hf), Angew. Chem. Int. Ed., 2021, DOI:10.1002/anie.202114303