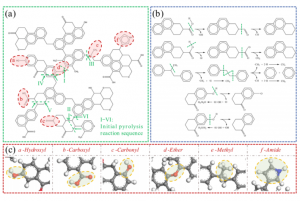
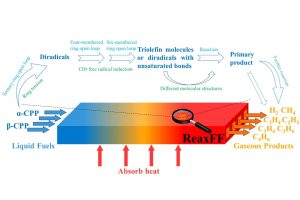
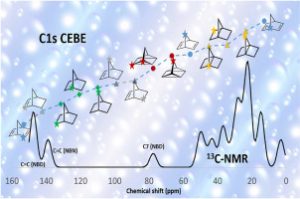
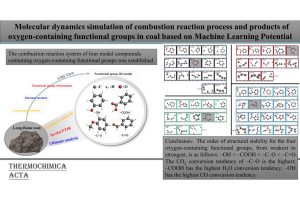
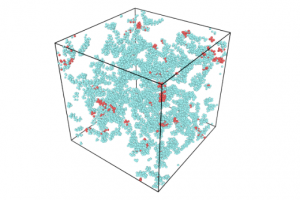
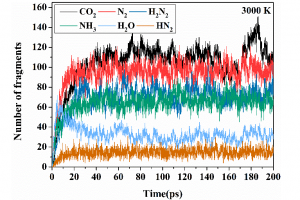
研究背景 在煤炭开采和加工过程中,甲烷和煤粉的混合爆炸对煤矿安全构成严重威胁。甲烷与煤粉的混合物易被机械摩擦、爆破等能量源引燃,其爆炸特性比单一燃料更复杂,产生的冲击波、火焰和有毒气体会加剧灾害。现有研究多聚焦宏观爆炸特性(如压力、火焰传播)和抑制技术,但对微观反应机制(如分子层面链式反应路径)缺乏深入探讨。本文通过实验和ReaxFF MD分子动力学模拟,结合烟煤(中国储量占比75%)的宏观爆炸行为与分子反应机理,旨在揭示混合爆炸的协同作用机制,为灾害防控提供理论依据。 研究方法 为了揭示甲烷/烟煤煤尘复合爆炸机理,甲烷/烟煤混合爆炸反应的分子模型构建过程。图1a为经典烟煤分子的初始结构,经几何优化和退火处理后获得稳定构型(图1b)。随后,利用 Packmol 按化学计量比将甲烷、氧气分子随机分布至周期性三维反应箱中,形成混合反应体系(图1c)。该模型结合了烟煤分子、甲烷与氧气的空间分布特性,密度设定为 0.5 g/cm³,为后续 AMS-ReaxFF-MD 模拟提供了原子级反应环境,旨在解析爆炸过程中分子链式反应路径与中间体演化机制。 图1 甲烷/烟煤煤粉复合反应系综分子模型构建 图2 褐煤分子的热解反应简化机制:(a)褐煤分子结构的初始热解反应序列;(b)二次热解过程中典型小气体分子(CO、CO2、CH4、H2O)的形成途径以及关键自由基(·H、·OH)的生成路径;(c)典型官能团 图3 甲烷/烟煤煤粉复合燃爆反应机制 主要研究结论 通过ReaxFF-MD模拟,分析了烟煤分子热解机理及主要气体组分和关键自由基的演化规律。结果表明,在预混体系中,烟煤分子有效地促进了燃烧反应过程,且在富氧水平较高时,促进作用更为显著.在甲烷/粉煤爆燃系统中,烟煤分子受热氧化分解,释放出大量的挥发分参与气相爆燃反应,该过程促进了混合体系的强度和加快了反应速率。 该研究为甲烷-氧气燃烧和甲烷/烟煤混合燃烧的反应途径提供了详细的见解。甲烷-氧气燃烧的主要途径包括CH4→ CH2CH3→ CH2O → CH2CHO → CO → CO2。此外,混合爆燃的甲烷和烟煤粉揭示了一个复杂的二级途径,导致形成各种副产物和中间产物,如CH4CH5O,CH4CH4O,CH4CH4O2,CH2O2,CH2CH2O2和CxHyO2。甲烷/烟煤爆燃的主要反应途径为CH4→ CH2CH3→ CH4O → CH2O → CH2CHO → CO+ H2→ CO2+ H2O。通过脱氢反应和结合转化维持了·H和·OH自由基之间的平衡,在混合爆燃和产物转化中起着至关重要的作用。 参考文献 Flame behaviors, pressure evolution, and molecular reaction mechanism of methane/pulverized bituminous coal hybrid deflagrations, Fuel […]