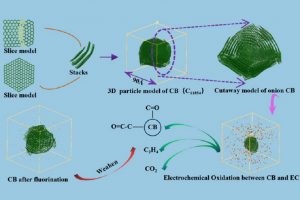
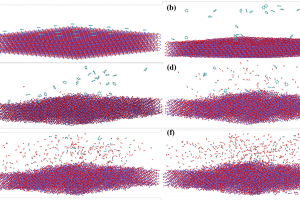
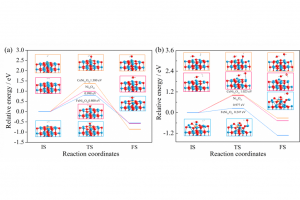
摘要 中南大学肖劲-仲奇凡课题组,通过高分辨率透射电子显微镜(HRTEM)、XRD、拉曼光谱和XPS实验检测,探讨了导电炭黑的结构特征。条纹的CB晶格长度大多<20Å,峰值约为10Å。条纹在0–360°处均匀分布,符合其“洋葱状”结构。总共堆叠了2-5层,平均堆叠数量为2.91,具有一定的顺序。弯曲条纹总长度的比例为67.13%,高于总数的比例(46.57%),表明弯曲条纹一般较长。然后通过构建CB(C11854)的洋葱状颗粒模型,结合FT-IR和XRD光谱计算验证了该模型的合理性。 作者同时在理论方面,采用基于反应力场的分子动力学(AMS软件ReaxFF模块)研究了CB和碳酸亚乙酯(EC)在锂离子电池中的电化学氧化行为。EC通过环内和环外O攻击CB,EC的分解产生CO2和C2H4。CB表面形成了各种O基团,EC和CB的破坏影响了电池的循环稳定性和寿命。在使用F2作为CB的保护后,EC的消耗减少,并且在CB表面仅形成少数O基团。这一结果为减缓CB和EC之间的电化学副反应提供了高精度的模拟支持。 扩展阅读: 中文详细解读:https://mp.weixin.qq.com/s/df7GchOCPdT_kVCdgKBNZw英文文献全文:Construction of a 3D “Onion-like” Model of Conductive Carbon Black for Lithium-Ion Batteries and Exploration of the Electrochemical Oxidation Mechanism of CB and Ethylene Carbonate via ReaxFF MD, Energy & Fuels. 2023, 37, 9, 6778–6790