概述
通过反应力场的分子动力学、蒙特卡洛方法,模拟燃烧、裂解、催化、超临界、电池、电气工程、建材、摩擦、生物药物、钙钛矿、半导体、含能材料、化学气相沉积等相关化学反应。包含多种化学反应加速算法;eReaxFF功能用于电池、太阳能电池等离子电子过程。笔记本即可运行一般几千原子的模拟,小规模集群可以支持上百万原子级别的模拟。
AMS 平台环境中 ReaxFF 的优势
- 图形界面完善,初学者也能正确使用。ReaxFF的开发者Adri van Duin在研究过程中,也大多使用AMS中的ReaxFF,详见Adir van Duin文章。
- 重写了ReaxFF全部代码,去除了内存方面的瓶颈,MPI结合OpenMP并行计算,效率远高于原始程序

- 算法更严谨,例如:
- 外加电场时,不仅对原子受力的影响,也考虑对能量。
- 原子间距过低时报错终止计算。
- 严谨检查力场参数,如果缺少参数则不允许运行。如果缺键参数仍然运行,结果是错误的。
- 化学反应发生的时间太晚,是ReaxFF模拟最常见的痛点,AMS中开发了加速反应发生的算法:
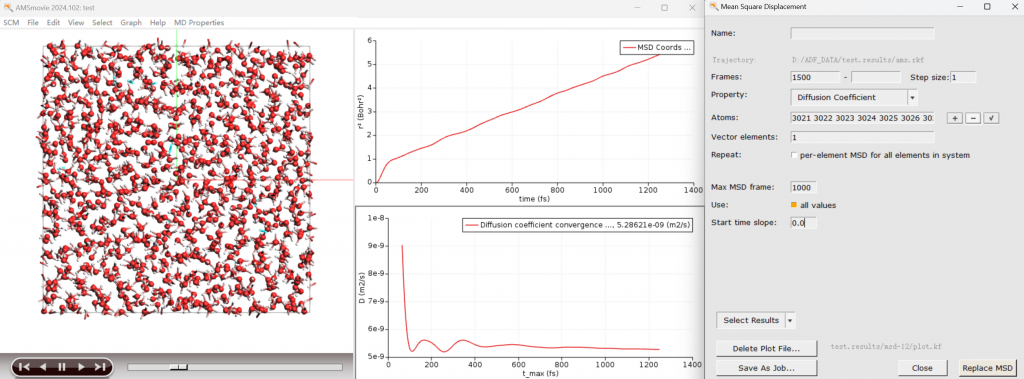
- AMS中包含大量围绕分子动力学研究化学反应的工具:
- 力场丰富,AMS2024 包括 133 种 ReaxFF 力场
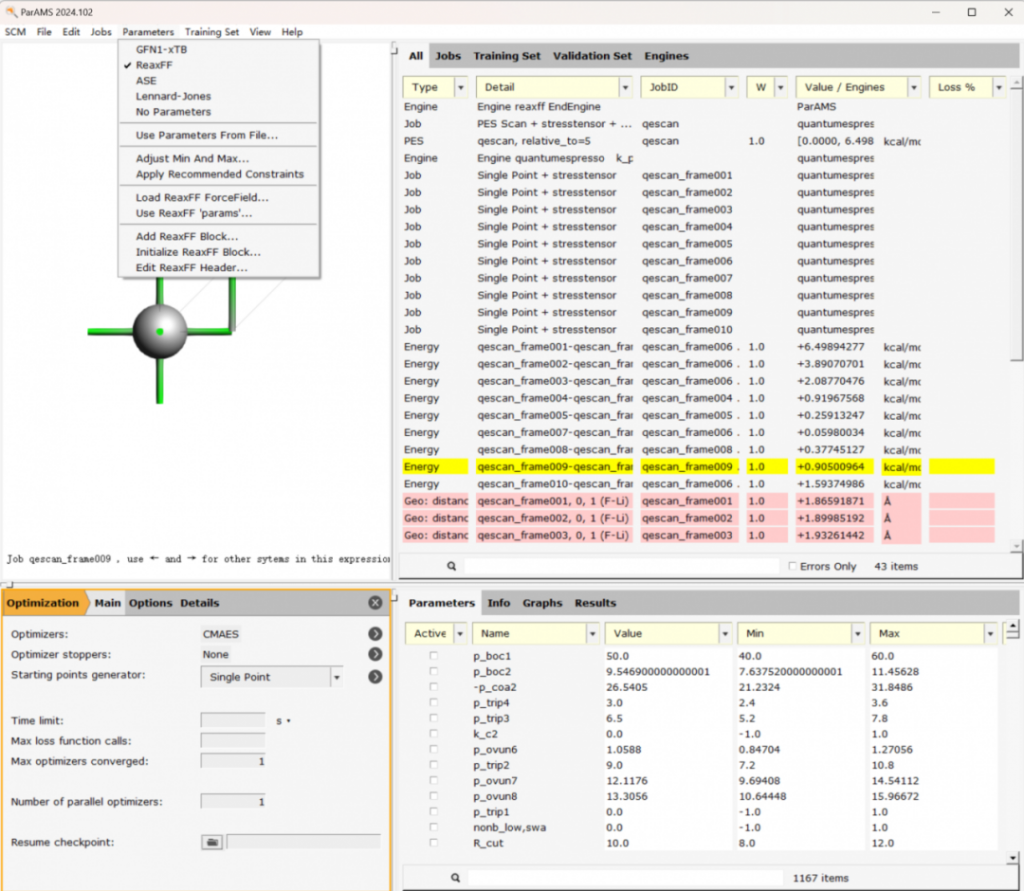
- AMS配套了力场训练工具
ReaxFF 功能列表
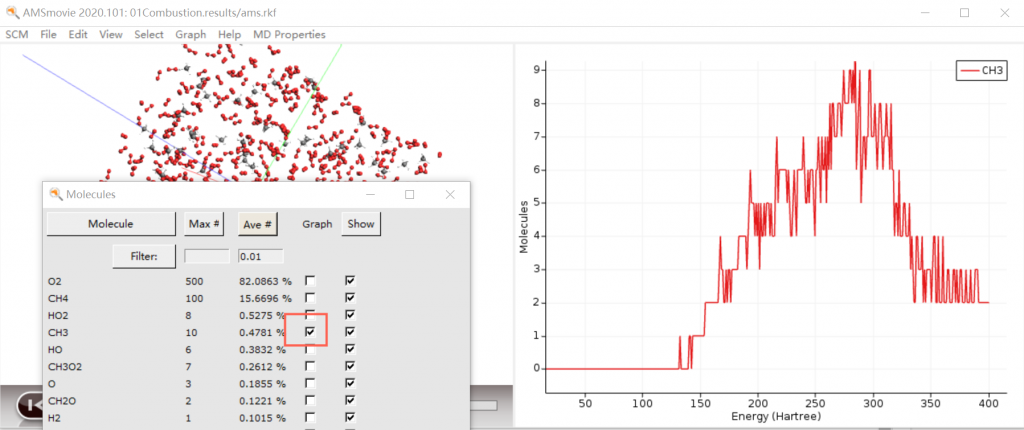
- 材料性质
对其他模块的依赖关系
- AdvancedWF模块
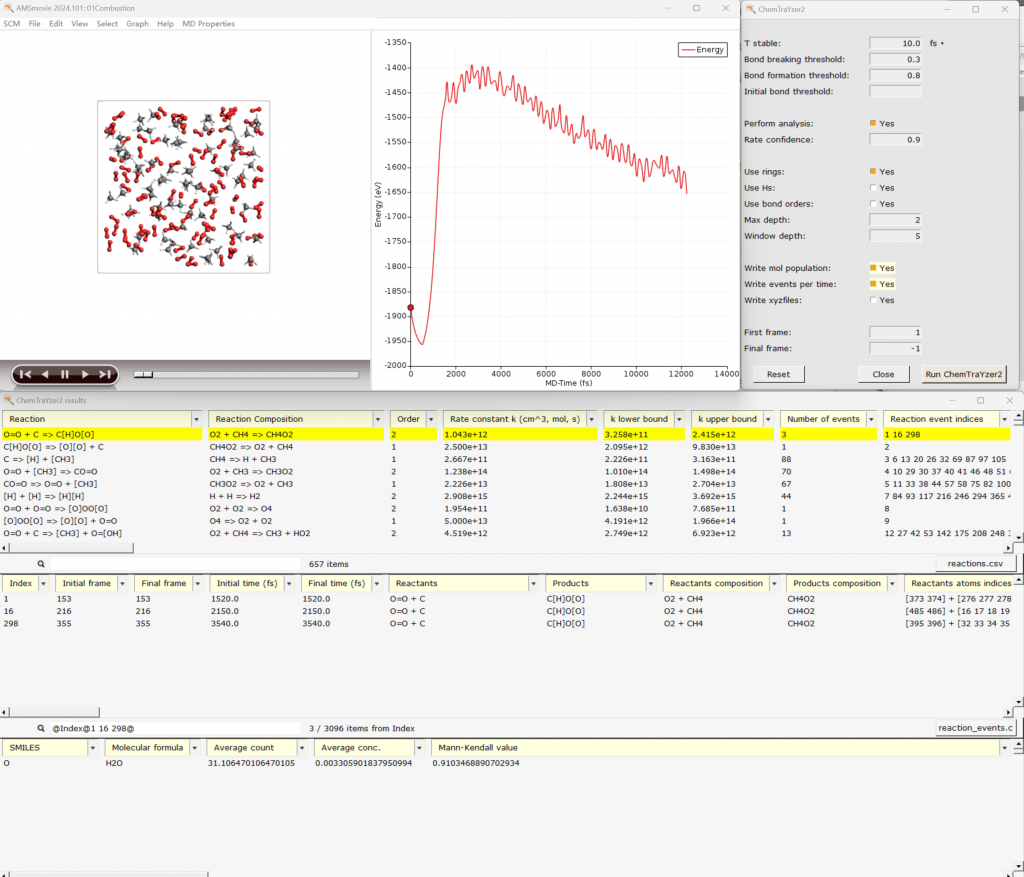
- ChemTraYzer2.0:对各类体系的轨迹进行基元反应,并能输出反应级别、实验单位的反应速率常数、布居统计等
- ACE-Reaction:反应通道的快速预测
- Reaction Discovery:快速确认(副)反应可能性、(副)产物可能性
- React Map:确定化学反应中反应物和产物之间的最佳原子映射
- ParAMS:力场优化、训练
- ADF或BAND模块:ADF与BAND是AMS内置的DFT计算模块,分别用于分子体系、晶体的DFT计算,生成DFT训练集,用于ParAMS进行力场的训练、优化