
对于某些特殊问题,我们希望人为地干预电荷分布,例如水分子解离为H+和OH–。使用DFT方法,自然地拉开H与OH的距离,我们期待解离的H原子上面没有电子,但随着拉伸的距离增大,H原子上,始终会出现电子。对于大的离子体系也是如此,有时候我们希望正负离子带有我们指定的电荷。常规的DFT计算,一般而言并不能达到这样的结果。
这种情况下,我们可以使用ADF中的FDE方法来实现。此处我们用H-OH来作为示范的例子,其他大的分子体系,计算流程是一样的。
1,对一个H+进行一次计算(此处泛函、基组均不起作用,因为没有电子,但其他的大分子的情况,则需要统一指定泛函、基组等),任务名此处定为H+.adf:
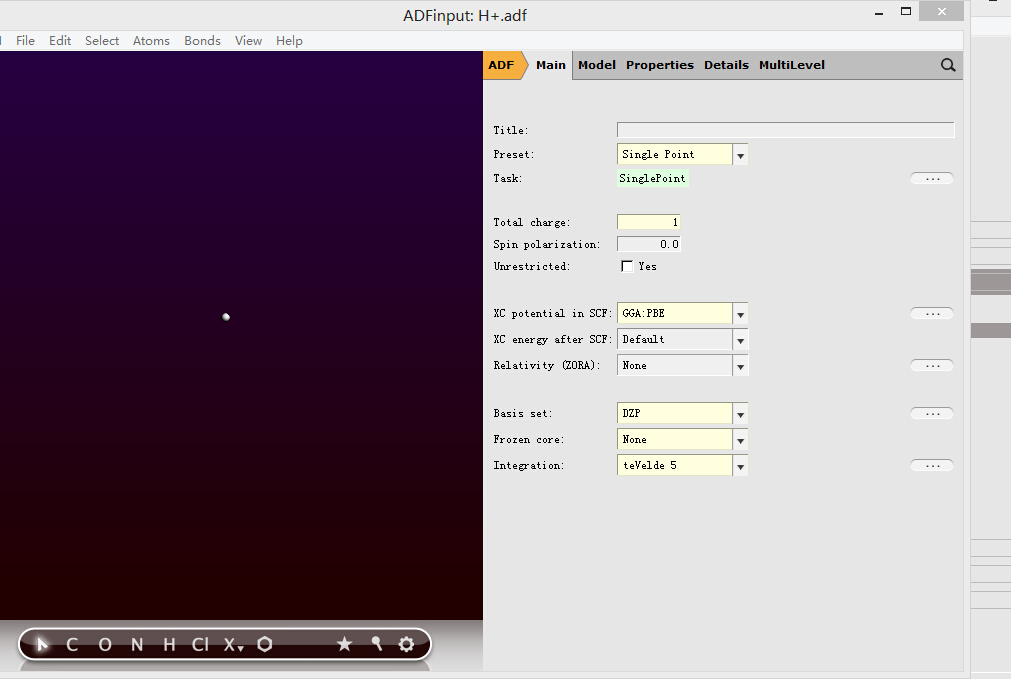
这一步计算得到H+.t21文件,保留此文件,下一步计算会使用;
2,计算质子逐步远离分子,例如该质子离羟基3.96埃的轨道与能量:
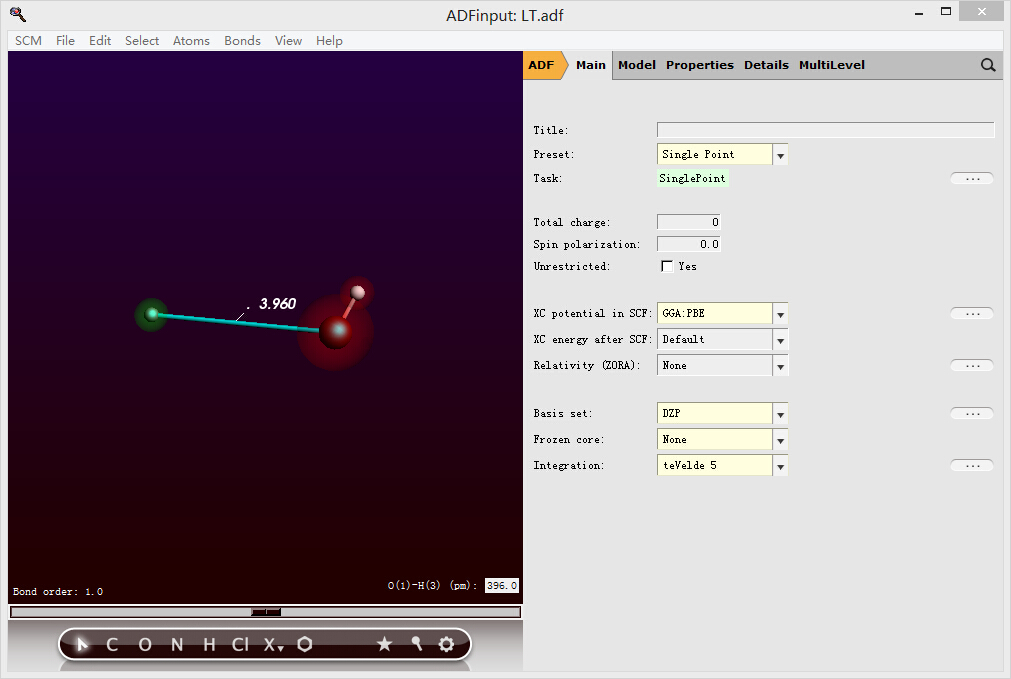
将质子和剩余部分分成两个区域(如图所示,表示为红色和绿色)——使用ADF的分区功能:
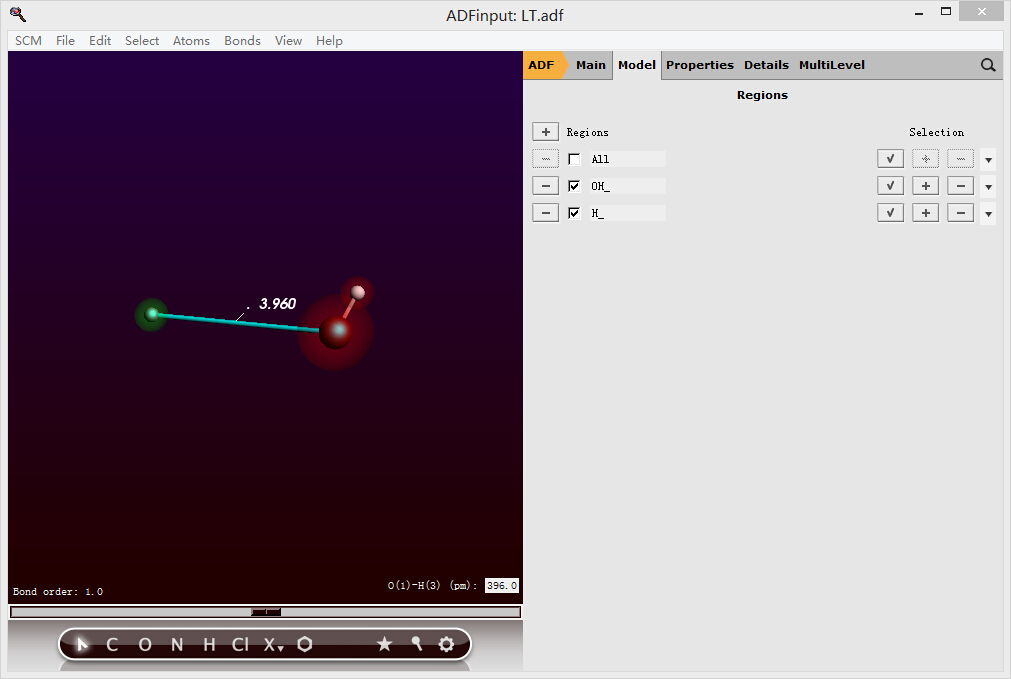
将质子设置为FDE区域:

同时使用上一步中得到的H+的t21文件,并分别在片段设置中设置两个片段的带电情况,并将“使用片段”的选项打钩,表示启用片段功能:
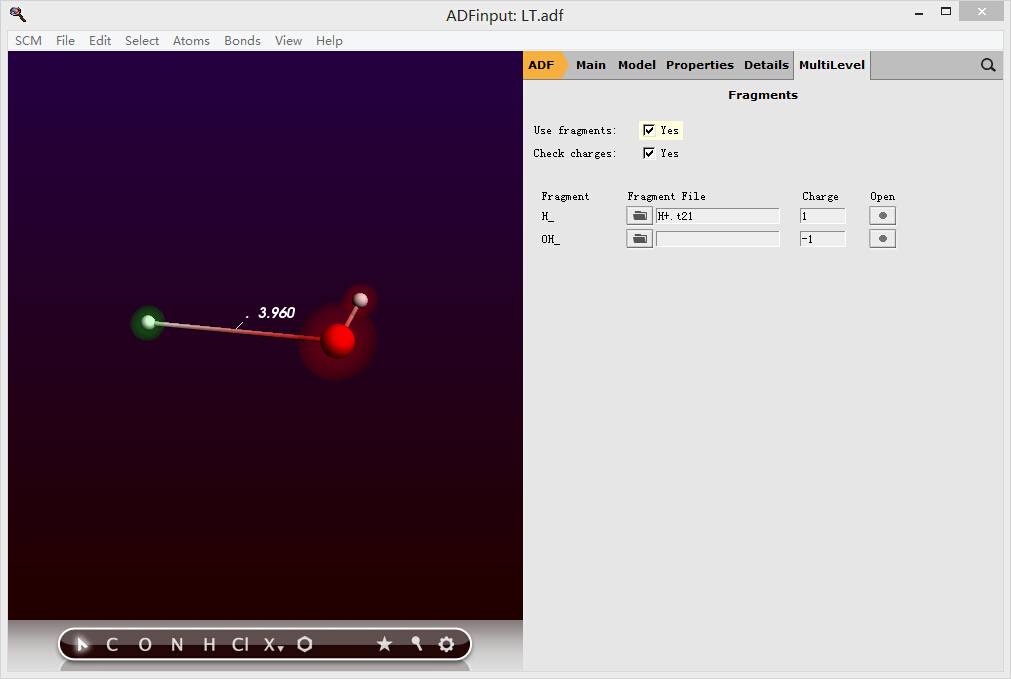
保存,并运行。其中保存的时候,弹出H+片段的结果是否覆盖,选No(实际上选Yes也无妨),运行;
3,结果查看: 在SCM-View窗口查看Mulliken电荷(注意此处H+上标记的Mulliken电荷为0,真正的电荷要加上FDE区本身的的电荷量+1):
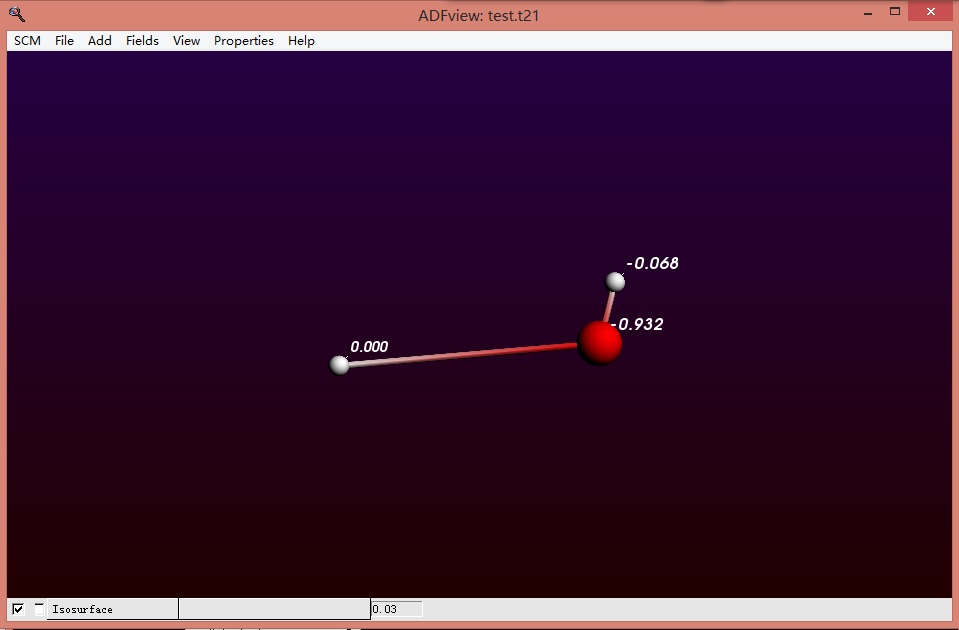
显示这样的计算结果,所有的电子确实分布在羟基上,质子上没有电子; 分别显示所有的电子轨道:
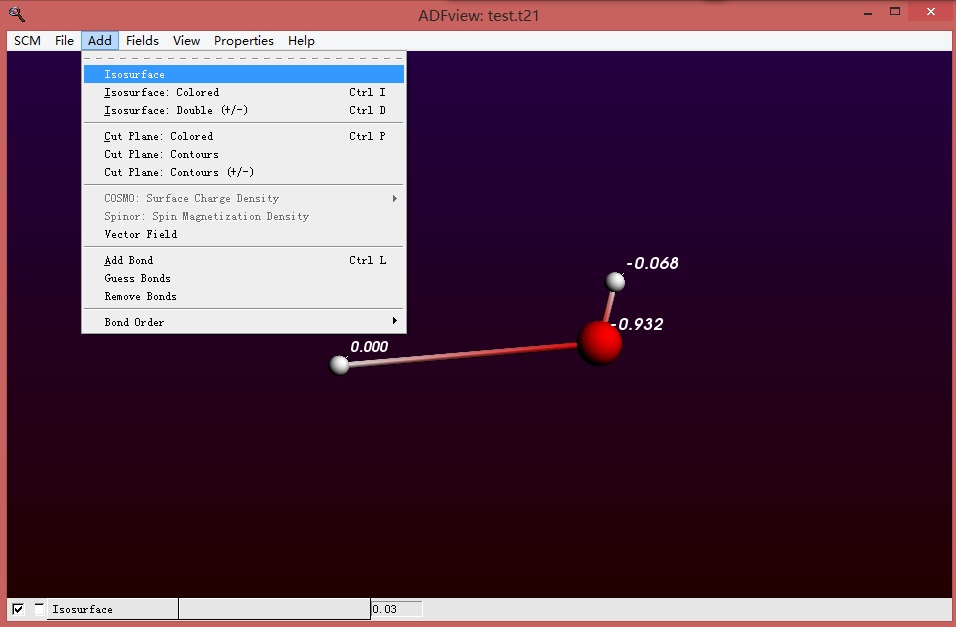
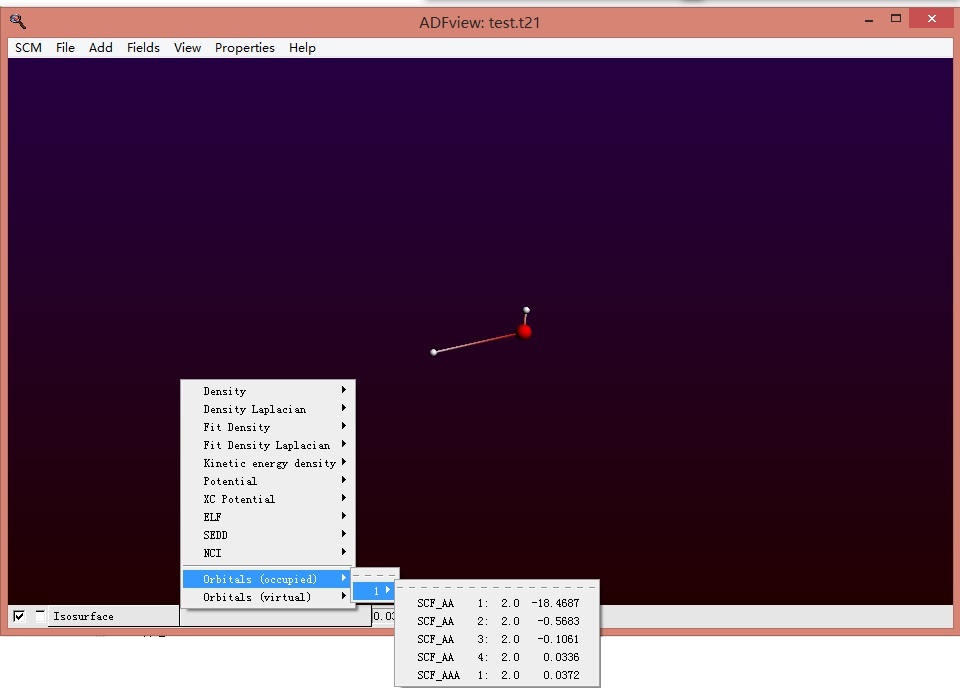
分别在orbitals(occupied)选中各个轨道,进而分别显示这些轨道,发现所有的电子轨道都分布在羟基上:
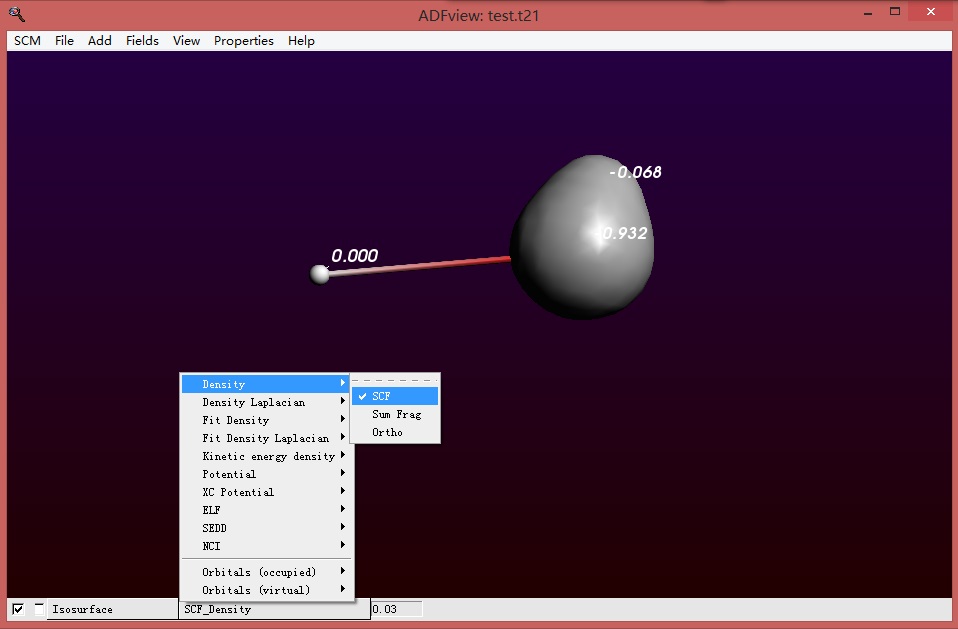
显示总的电子密度空间分布,也可以看见所有电子都分布在OH–上面:
 到此完成。
到此完成。
更多案例参见费米科技WIKI-ADF
版权声明:本文版权归属费米科技,未经许可,任何其他单位不得用于商业、市场活动。