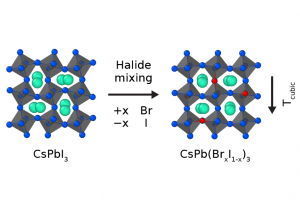
摘要 金属卤化物钙钛矿已成为光电应用中极具前景的材料,其卓越的性能使其成为太阳能电池和LED的理想候选材料。现有的钙钛矿组合物通常涉及不同离子的混合,目的是针对特定应用,对材料的光电性质和稳定性进行微调。为了理解离子混合的原子效应,埃因霍温理工大学和宾夕法尼亚州立大学的研究人员开发了一种用于大规模分子动力学模拟的无机金属卤化物钙钛矿(CsPbX3,X=Br 或 I)的 ReaxFF 力场[1]。 研究人员利用 DFT 计算生成的参考数据集,使用 AMS 软件中的 ParAMS 参数化功能,基于先前开发的 CsPbI3 的力场(该力场有助于研究体材料降解反应[2]以及表面和晶界的影响[3]),对该力场进行了扩展, 得到了新的无机卤化物钙钛矿的 ReaxFF 力场[4] CsPb(BrxI1-x)3 ,新开发的 I/Br/Pb/Cs 参数集首次对 Br 进行描述。在包括状态方程、混合焓、降解反应和缺陷迁移能垒在内的各种基准测试中,该力场表现良好。在分子动力学模拟中,该力场可以准确地再现材料的有限温度效应,例如无机钙钛矿的各种体相之间的相变。 使用新的力场参数,研究人员确定,由于 I 和 Br 离子的尺寸失配,卤化物混合对材料中八面体的相变温度和倾斜动力学有着深远的影响。利用卤化物混合的稀释极限(即取代钙钛矿晶格中的单个卤化物),确保这种效应是非局部的,距离混合位点高达两纳米。混合效应的非局部性,解释了为什么少量卤化物混合会对材料性质(如相变温度)产生很大影响。新的 ReaxFF 力场参数,为大型现实体系通过原子模拟,进一步探索无机混合卤化物钙钛矿的复杂动力学铺平了道路。 参考文献 Pols, M.; van Duin, A. C. T.; Calero, S.; Tao, S. Mixing I and Br in Inorganic Perovskites: Atomistic Insights from Reactive Molecular […]