
荷兰自由大学F. Matthias Bickelhaupt与Trevor A. Hamlin课题组最近提出了基于活化畸变模型(Activation Strain Model, ASM)的化学反应性研究方法,基于该模型的化学反应分析工具PyFrag 2019发布于材料化学模拟平台Amsterdam Modeling Suite(简称AMS) 2019.3版中。这种最先进的计算技术能够协助化学家们预测化学反应性,并合理设计新的化学反应,普适于所有反应类型,包括无机、有机、超分子、生化等领域。其他方法只分析势能面上某一个驻点,而该方法对反应坐标沿路能量的变化进行分析。
ASM方法基于ADF模块(或BAND模块)非常经典的片段分析功能,主要基于:
- 反应物分子从单独存在时的能量最低结构,形变到反应要求的结构所需的能量
- 变形的反应物分子间的相互作用能
这种方法让用户能够洞察决定反应能垒的诸多要素,以及化学反应性趋势。ASM虽然已被多篇综述介绍(Chem. Soc. Rev. 43, 4953, 2014;WIREs Comput. Mol. Sci. 5, 324, 2015;Angew. Chem. 129, 10204, 2017),但尚缺乏具体的清晰的使用步骤指南,因此作者在本文中以AMS软件中的ADF模块为例,介绍ASM的应用步骤。对于周期性体系而言,该方法同样适用,只是对应使用AMS软件中的BAND模块。之所以使用AMS软件,是因为该软件支持:
- 与ASM匹配的能量分解分析(EDA):将ASM得到相互作用能分解为几项具有明确物理含义的能量项(具体含义参考:能量分解分析(EDA),或本文介绍文献的原文)
- 分子轨道分析:将分解得到的各能量项与反应物分子轨道联系起来
路线图如下:
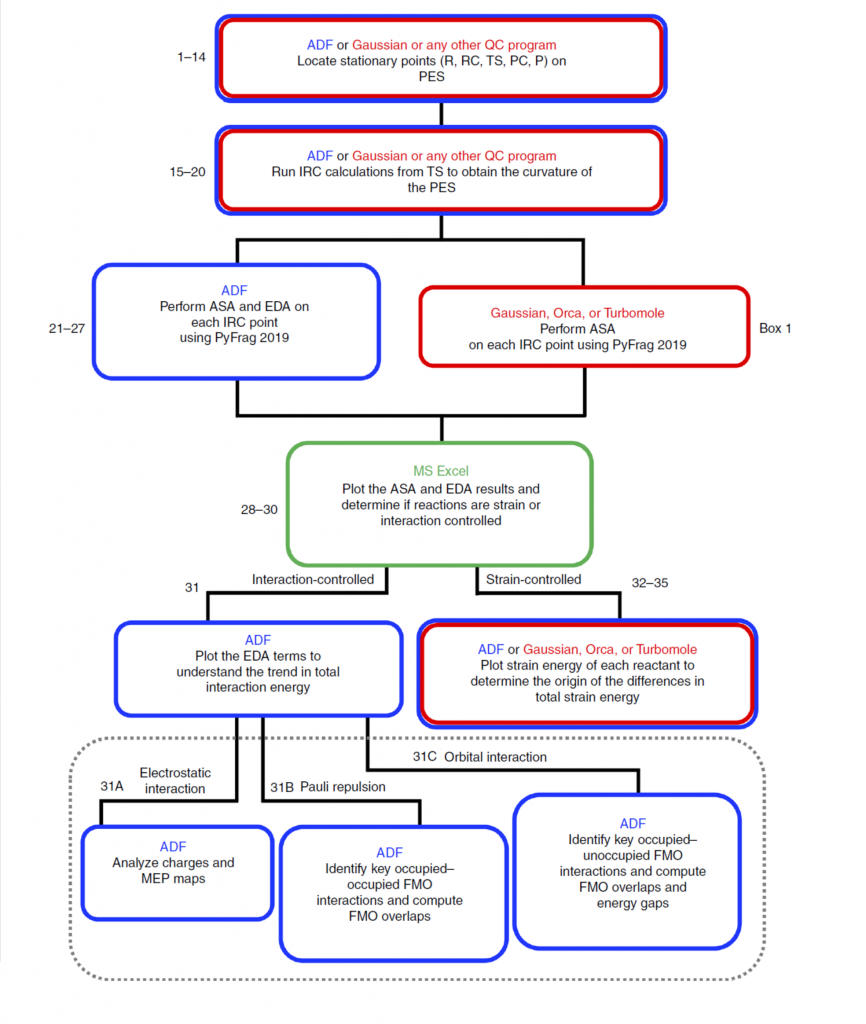
以甲基叠氮与环庚炔、环壬炔的反应为例:
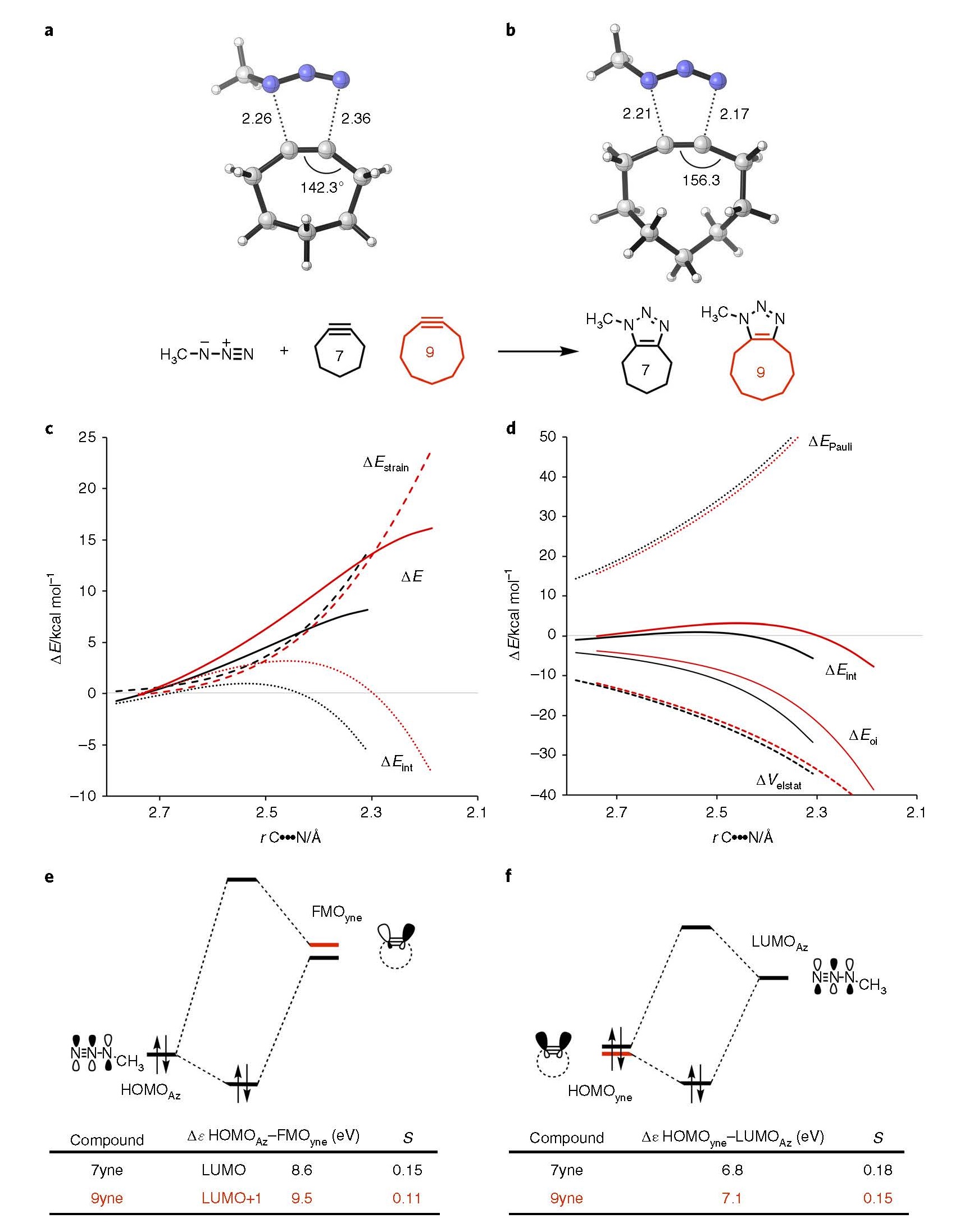
(a)、(b) 环庚炔、环壬炔与甲基叠氮反应过渡态结构;(c) 体系的形变能、分子间相互作用能,以及二者之和(也就是总能)的变化曲线;(d) EDA分析分子间相互作用各能量项变化曲线; (e)、甲基叠氮HOMO与环炔碎片LUMO之间的相互作用;(f)甲基叠氮LUMO与环炔碎片HOMO之间的相互作用。S为轨道重叠积分,r为距离
上图中ASM-EDA分析中得到的能量项,被投影到新形成的C····N键平均距离。该距离是这一类反应的关键反应坐标,直接与反应过程相关。得到的活化应变图表明,环壬炔的环加成势垒比环庚炔高。由于二者的形变能ΔEstrain曲线几乎重叠,因此相互作用能ΔEint的大小趋势决定了反应能垒的大小趋势。进一步分析ΔEint各分项的趋势,发现泡利排斥能或多或少被静电相互作用能ΔEelstat抵消(上图d)。另一方面,轨道相互作用ΔEoi对总的相互作用能趋势,即反应能垒的趋势,起到决定性作用。
Kohn-Sham分子轨道分析印证了ΔEoi趋势的合理性:环庚炔、环壬炔,何者的预形变或弯折越大,则其的FMO(碎片分子轨道) Gap就越小,同时空-占轨道重叠越强(注意这里的空、占轨道是指发生相互作用的分子轨道,一定是某个分子的占据轨道与另一个分子的空轨道),越利于反应。与环壬炔相比,环庚炔的形变程度更大,导致其HOMO稳定性更低(从而能量越高,反之亦然),LUMO稳定性更高,从而导致FMO Gap更小。另外,环庚炔形变更大,从而导致其HOMO、LUMO朝向甲基叠氮部分与甲基叠氮LUMO、HOMO的重叠比环壬炔更大。所有这些因素叠加在一起,导致环炔越小,轨道相互作用越强。
PyFrag 2019使用手册,参考:https://www.scm.com/doc/ADF/Input/PyFrag.html