AMS:专业的材料与化学模拟平台
适合计算化学初学者、实验工作者入手的专业软件
多尺度计算模拟体系
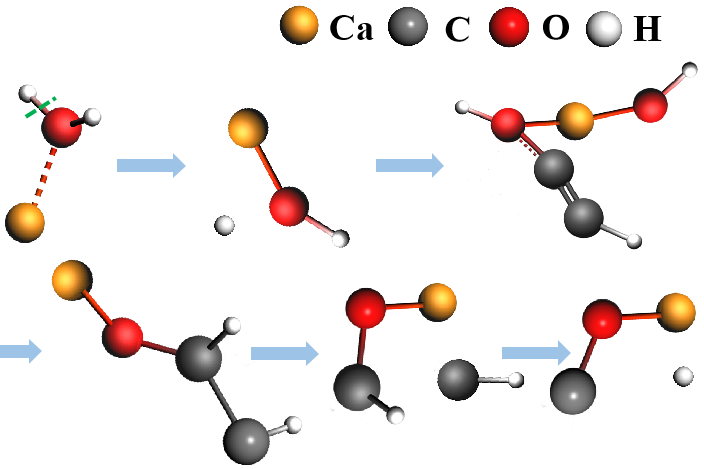
- 分子体系的量子化学计算:化学反应机理研究、丰富的光谱性质预测、发光、热力学性质、化学键的机理研究、重元素配合物与团簇、多金属氧酸盐、分子构象搜索等
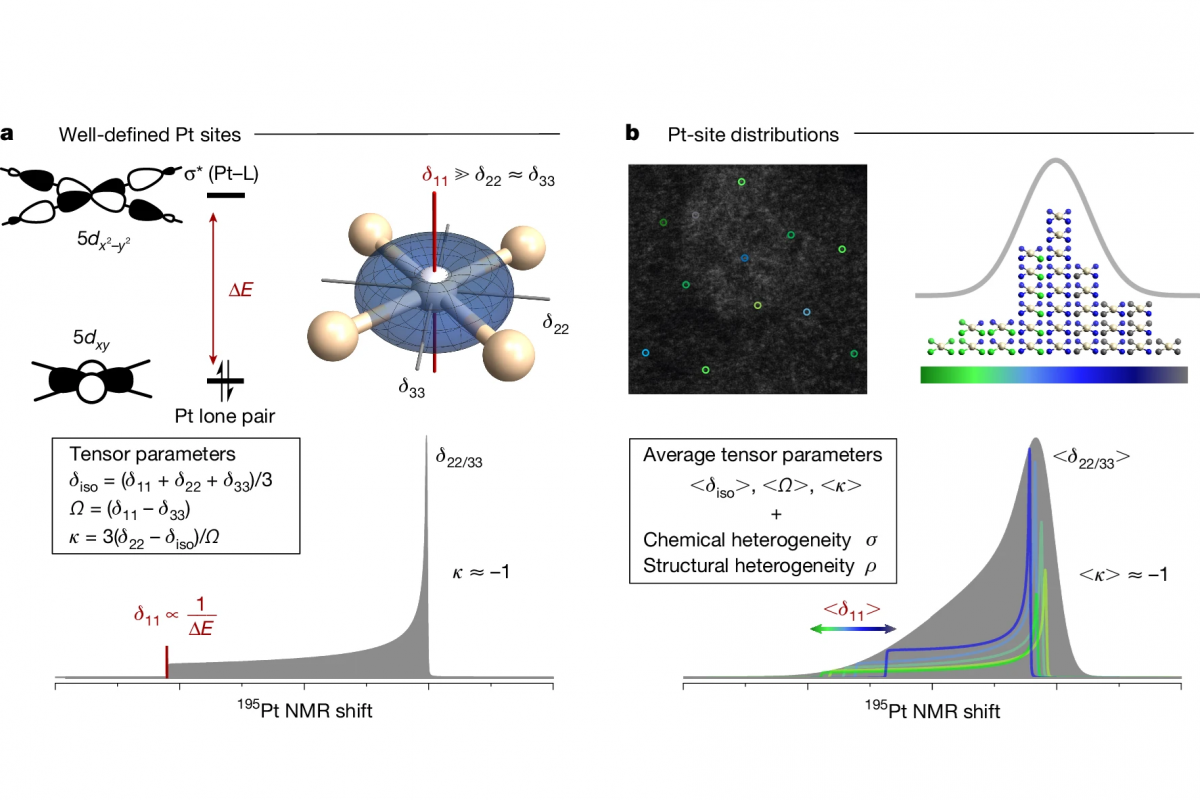
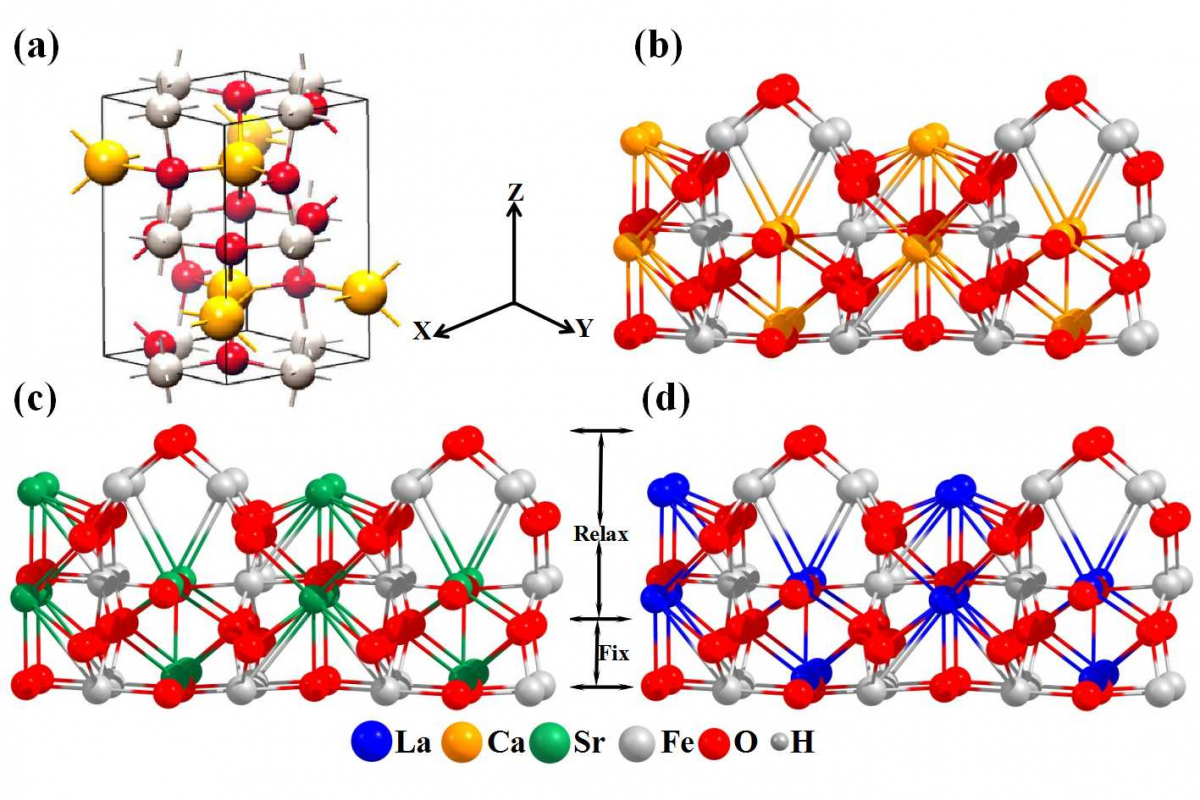
- 晶体、低维材料:吸附、表面催化、单原子催化、化学反应机理、磁性质、材料电子学性质、化学键的机理研究、力学性能、热力学性质、光的吸收与折射等
- 宏观物质:离子液体、溶液、气液平衡、液液平衡、共晶、低共熔溶剂筛选等,广泛应用于化工、制药领域的研究
友好的图形界面操作
- 图形界面操作,即可完成建模、计算、作业管理、结果分析、图谱展示
完善、全面的计算模拟方法
- 分子动力学:基于经典力场、反应力场、机器学习势、半经验量子化学方法的分子动力学与蒙特卡洛模拟,被广泛地应用于化学反应机理与预测研究,以及微细加工、燃烧、热解、催化,以及半导体、聚合物物性的研究
- 蒙特卡洛:巨正则系综蒙特卡洛、Force Biased 蒙特卡洛、动力学蒙特卡洛
- QMMM等多尺度模拟方法,结合不同计算引擎用于大体系计算
- 接近化学工业研究实际:微观动力学用微观机理,预测宏观化工过程
完善的中文教程系统
- 初学者可以顺利通过中文教程,正确掌握软件








平台概况
精确、智能化、集成化、学生友好易用的图形化计算化学平台
- 图形窗口与计算:支持最新Win、Linux、Mac系统
- 材料数据库:内置分子与晶体结构数据库、流体热力学分子库
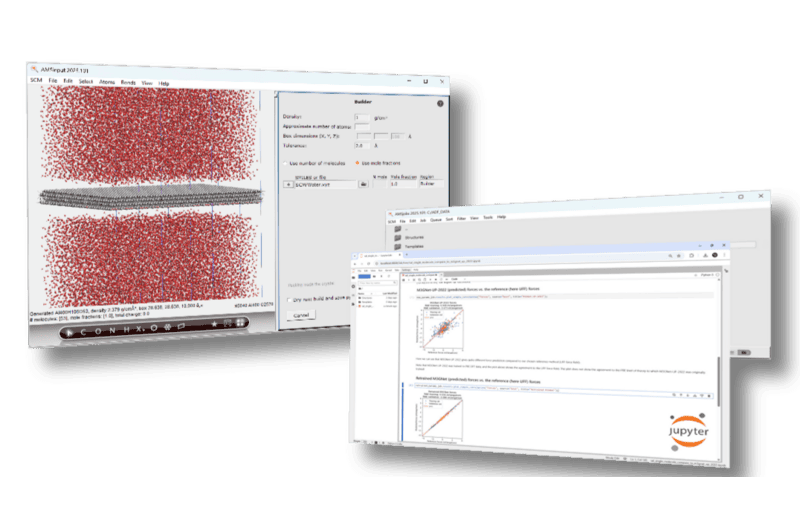
- 完善的建模工具:智能化操作方式,构建晶体、分子、表面、界面、聚合物、均相与非均相混合物模型,快捷构建吸附、化学反应等多种复杂模型
- 数据分析与作图:快速数据处理,与图谱展示,支持导出数据、图标
- 脚本与作业管理:图形化设置生成计算脚本、批处理作业,支持作业模板定制,灵活提交计算到远程服务器,支持所有常见的大型集群的管理系统
- 高度并行化计算:跨节点高效并行化计算,最大程度提高研究效率
- 智能化参数检测:自动检测不合理、错误参数,并提供建议参数
- AMS驱动环境:调用各个功能模块进行多尺度模拟、分子动力学模拟、巨正则系综蒙特卡洛模拟、动力学蒙特卡洛模拟等
- 参数开发环境:整合DFTB参数与ReaxFF力场开发工具,图形化创建训练集、导入样本、训练参数、生成文件、检验评估参数
AMS 中的功能模块
多尺度、多模型、多功能的建模和计算与分析
BAND & Quantum Espresso
BAND:1D、2D体系、杂化泛函比平面波快1-2数量级,高效表面化学过渡态计算,表面吸附化学键分析;
Quantum Espresso:基于平面波/赝势方法的DFT计算,结构计算、能带与态密度、磁性、表面化学等功能
了解详情...
Advanced Workflows & Tools
分子动力学辅助工具包:分析基元反应、反应速率常数,ReaxFF力场训练,AIMD机器学习,微观动力学,动力学蒙特卡洛,OLED 沉积模拟
了解详情...
ForceField & ML Pot
AIMNET2、通用机器学习势M3GNET、CHGNet,经典力场GFN-FF、UFF、UFF4MOF、UFF4MOF-II、GAFF(包含几乎所有元素)
了解详情...
DFTB & MOPAC
近似量子化学方法,包括SCM自行开发DFTB参数,覆盖了几乎元素周期表2/3的元素,支持分子动力学、能带与态密度、结构预测、大分子光谱等
了解详情...
Bumblebee - OLED/OFET 器件模拟
输入分子材料微观性质,使用三维蒙特卡洛方法,模拟像素点的不同堆叠结构,获得电流电压特性、激子传输与光电过程、载流子迁移,研究像素的发光特性、效率、退化
了解详情...
费米科技作为荷兰 SCM 公司在中国唯一合作伙伴,负责 AMS 中国销售与售后技术支持
欢迎试用最新版本的AMS
试用与购买
欢迎试用最新版本的AMS