
目录
使用块体材料DFT计算数据优化ReaxFF力场
提示:下面的操作,除了DFT计算之外,其他只在Linux、Mac系统测试过。本教程仅适用于2019.1以下版本。
ADF软件集成了使用蒙特卡洛方法优化力场参数的脚本。目前已经可以较为方便的使用,脚本经过测试,可以正常工作,但大都是在命令行下工作。用“优化”的方式,也可以创建一些新的力场:一般在相似力场的基础上,对力场中的元素进行修改,使用DFT计算这些新元素体系的一些结构的能量,然后使用MCFF对力场参数进行优化。
工作过程分为如下几步:
- 可以调整键长键角、晶格常数等参数,得到不同的结构,也可以通过MD过程得到一系列代表性的结构。使用DFT方法计算这一系列结构的能量;
- 通过DFT计算的结果,整理出geo、trainset.in文件;
- 准备ffield文件:该文件是力场的初始猜测(如果是要创建新力场可以找到相似力场,直接修改元素符号得到),如果是优化现有的力场(例如CHO.ff),可以直接拷贝过来,改名为ffield即可,参考:力场文件在哪里?
- 使用脚本自动生成ffield_bool文件,并可以进行修改,这个文件是定义哪些参数需要优化,其中为0的位置,表示ffield这个位置的参数不需要优化,里面的值只有0和1两种,1表示需要优化。
- 设定力场文件里面,需要优化的参数的数值的区间:ffield_max、ffield_min这两个文件与ffield格式一致,只是分别给出的是优化力场的时候,ffield中对应的参数的可能的上限和下限。优化过程中,程序会不断调整力场里面需要优化的参数的数值,但这些参数的数值都将保持在这个上下限区间内。区间需要设置的尽量合理一些,可以提供成功率,提高力场的质量;
- mcff.run文件可以直接拷贝过去使用,但需要注意修改几个参数:
- mcbeta=1/KbT。注意该温度和实际ReaxFF模拟时使用的温度没有关系,只是参数空间的“温度”;
- mcdbet是蒙特卡洛逐渐降温的速率(每步mcbeta增大多少);
- mcffit是进行多少步蒙特卡洛拟合;
- mcmxst是允许运行的最大步数,这个参数需要改大,否则默认值为0,只运行一步。
本文以石墨烯片层为例,DFT计算得到的层间距为3.355埃(如下图中所示,单胞内的两层石墨烯):

而CHO.ff力场优化得到的间距是3.10埃。
本文尝试通过优化CHO.ff的参数,使得新的CHO.ff力场也能优化出与DFT接近的间距。因此DFT计算主要围绕这一点,计算了平衡间距附近的一些结构的能量,用这些计算结果优化CHO.ff力场。如果用户关心其他方面,例如其他键长,也可以用DFT计算类似的计算。ReaxFF中有的晶体常数计算不准确,也可以通过调整不同的晶格常数来计算DFT能量,并以此优化对应的力场。
注意
下文生成的geo、trainset.in、ffield、ffield_min、ffield_max、ffield_bool、ffield、mcff.run文件必须在同一个目录下,优化力场的过程也是通过命令行,在该文件夹进行。因此要确保在该文件夹下,可以直接运行*.run,并且该文件夹的完整路径中不包含中文字符、空格等。
Windows中的运行命令行的方法:
- 双击运行adf201*.*/adf_command_line.bat打开命令行
- 输入sh或bash回车,之后就可以像Linux一样使用脚本了
- 例如下文的第一个命令,需要输入:
$ADFBIN/startpython LT_to_trainset.py *.t21
后面其他的命令,也需要在前面加${$ADFBIN/}$,但是像cp、chmod这样的命令,并不是ADF的命令,而是系统的基本命令,前面不需要加${$ADFBIN/}$,用户需要自行尝试。
1. 一系列结构的DFT计算
这里我们使用ADF软件中,用于计算周期性体系DFT计算的BAND模块进行计算。改变两层石墨烯之间的距离,分别为3.913、3.718、3.416、3.355、2.992、2.618、2.316埃,计算了他们的能量。如下图所示:

说明:
这里只是为了演示过程,因此DFT计算的时候,没有设置很精确的参数,用户应该根据自己的体系,选择适合的基组、泛函,具体可以参考:不同泛函的特点、ADF:一般情况下如何选择基组Basis Set、diffbasisfordiffelement,另外因为不关心能带等性质,因此可以设置k点为1 1 1,也就是只计算Gamma点(ADFinput > Details > Numerical Quality > k-space : Gamma Only),这样可以大大节省计算量,而不影响力场优化的可靠程度。
计算结束后,在*.logfile尾部可以看到以三种单位显示的形成能:

我们需要其中单位为kcal/mol的数值,例如该结构能量为-544.6622 kcal/mol。
注意
能量是一个相对值,BAND直接给出形成能,对同一个体系(原子相同),形成能可以代替总能量。
2. 将DFT计算结果、晶体结构信息导入到geo文件
geo文件的格式:
BIOGRF 200 DESCRP 1_reax REMARK Created by ADFinput RUTYPE SINGLE POINT CRYSTX 2.46000 2.46000 6.71000 90.00000 90.00000 120.00000 HETATM 1 C 0.00000 0.00000 1.67750 C 1 1 0.0 HETATM 2 C 0.00000 0.00000 -1.67750 C 1 1 0.0 HETATM 3 C 0.71014 -1.23000 1.67750 C 1 1 0.0 HETATM 4 C -0.71014 1.23000 -1.67750 C 1 1 0.0 UNIT ENERGY kcalmol ENERGY -596.8255 END …… BIOGRF 200 DESCRP 3_reax REMARK Created by ADFinput RUTYPE SINGLE POINT CRYSTX 2.46000 2.46000 6.71000 90.00000 90.00000 120.00000 HETATM 1 C 0.00000 0.00000 0.63850 C 1 1 0.0 HETATM 2 C 0.00000 0.00000 -1.67750 C 1 1 0.0 HETATM 3 C 0.71014 -1.23000 0.63850 C 1 1 0.0 HETATM 4 C -0.71014 1.23000 -1.67750 C 1 1 0.0 UNIT ENERGY kcalmol ENERGY -544.6622 END
里面有晶体结构信息、能量信息等,手写起来非常麻烦。我们可以通过如下操作,快捷生成该信息:
- 创建文本文件geo。
- 在完成该结构的BAND模块的DFT计算之后,直接切换到ReaxFF模块,然后另存一下任务,例如命名为2.316_reax。忽略弹出的“没有设置力场”的提示,直接保存。保存完毕之后,查看生成的2.316_reax.run文件中,包含这部分信息的大部分内容:
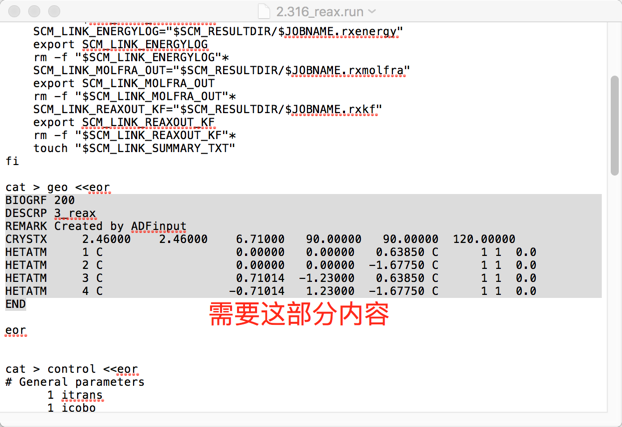 将这部分内容复制到geo中。
将这部分内容复制到geo中。 - 参考上面所示的geo的格式,略作修改:1)添加能量信息;2)插入一行:“UNIT ENERGY kcalmol”;3)插入一行“RUTYPE SINGLE POINT”(这一行不插入也可以,用于说明数据来源)
- 将所有DFT计算的结果,类似地添加到geo文件中。

3. 生成trainset.in文件
这个文件并不是geo文件相同的作用,而是对力场优化的过程进行一些权重设置,也就是用户可能特别要求某些参数更重要,权重就可以设置的很高,这样在优化力场过程中,就会更大程度地满足该参数条件。
这个文件最多包含5个部分(详细说明,参考英文手册 page 27),包括CHARGE、HEATFO、GEOMETRY、CELL PARAMETER、ENERGY。例如关于原子电荷的:
CHARGE #Iden Weight Atom Lit chexane 0.1 1 -0.15 ENDCHARGE
表示geo文件里面叫做chexane的那个结构,第1个原子的电荷值(来自文献或DFT计算,其中Lit是“文献”的意思,不过这一行以#开头,只是注释,并不影响计算)为-0.15,优化时权重为0.1(权重之和不需要等于1,程序会自动归一化)。如果权重很大,那么在优化力场的过程中,将努力保证新力场能符合该要求。不需要五个部分全部设置,设置几个部分,就生效几个部分。
我们可以用脚本自动生成一个trainset.in文件,有必要的话,在它的基础上修改。在命令行执行:
adfreport geo -rxtrainset > trainset.in
即可生成trainset.in文件。其中adfreport是ADF软件内置命令。
本例生成的trainset.in文件:
CHARGE 1_reax 0.1 1 0.0 1_reax 0.1 2 0.0 1_reax 0.1 3 0.0 1_reax 0.1 4 0.0 3_reax 0.1 1 0.0 3_reax 0.1 2 0.0 3_reax 0.1 3 0.0 3_reax 0.1 4 0.0 2_reax 0.1 1 0.0 2_reax 0.1 2 0.0 2_reax 0.1 3 0.0 2_reax 0.1 4 0.0 2.5_reax 0.1 1 0.0 2.5_reax 0.1 2 0.0 2.5_reax 0.1 3 0.0 2.5_reax 0.1 4 0.0 4_reax 0.1 1 0.0 4_reax 0.1 2 0.0 4_reax 0.1 3 0.0 4_reax 0.1 4 0.0 5_reax 0.1 1 0.0 5_reax 0.1 2 0.0 5_reax 0.1 3 0.0 5_reax 0.1 4 0.0 6_reax 0.1 1 0.0 6_reax 0.1 2 0.0 6_reax 0.1 3 0.0 6_reax 0.1 4 0.0 ENDCHARGE GEOMETRY 1_reax 0.1 1 3 1.420281246655042 1_reax 0.1 2 4 1.420281246655042 3_reax 0.1 1 3 1.420281246655042 3_reax 0.1 2 4 1.420281246655042 2_reax 0.1 1 3 1.420281246655042 2_reax 0.1 2 4 1.420281246655042 2.5_reax 0.1 1 3 1.420281246655042 2.5_reax 0.1 2 4 1.420281246655042 4_reax 0.1 1 3 1.420281246655042 4_reax 0.1 2 4 1.420281246655042 5_reax 0.1 1 3 1.420281246655042 5_reax 0.1 2 4 1.420281246655042 6_reax 0.1 1 3 1.420281246655042 6_reax 0.1 2 4 1.420281246655042 ENDGEOMETRY
可以看到7个结构中,每个结构的4个原子,电荷都是0。
1_reax 0.1 1 0.0
表示结构名为1_reax的那个结构里面第1个原子的电荷为0,这一条要求的权重为0.1(当然后面的其他权重也都是0.1,不过对石墨烯而言,电荷为0是比较符合实际情况的,因此步需要修改)。
另外,GEOMETRY这一块,例如:
1_reax 0.1 1 3 1.420281246655042
表示1_reax这个结构第1、3原子之间键长为1.420281246655042埃,这实际上就是六元环的键长,这个我们并不关心,也不想去优化它,因此也可以要求优化力场的过程中,保持这一点不变。权重默认值也是0.1。
对本例而言,没有需要修改的地方。如果一定要修改,可以直接将DFT优化得到的层间距(也就是第1、2原子间距离)写入到这个文件里面:
1_reax 1.0 1 2 3.355
第一个结构1_reax就是DFT优化的最优结构。可以设置这个权重非常大,例如是其他条件的权重的10倍(即1.0)。但本例中,实际上并没有这样做。使用了默认的trainset.in文件。
Trainset.in文件格式,参考:https://www.scm.com/doc/ReaxFF/trainset_descrp.html
4. 准备ffield、ffield_min、ffield_max、ffield_bool文件
生成ffield
本例中,我们可以直接在CHO.ff的基础上修改,所以直接将其拷贝过来,改名为ffield即可。
生成ffield_min、ffield_max
ffield的格式、参数的具体含义,参考英文手册(page 16)。
我们这里可以只优化:
1 1 156.5953 100.0397 80.0000 -0.8157 -0.4591 1.0000 37.7369 0.4235 0.4527 -0.1000 9.2605 1.0000 -0.0750 6.8316 1.0000 0.0000
这部分的四个键能:156.5953 100.0397 80.0000 -0.8157 其中“1 1”表示这一部分参数,是关于第一个元素和第一个元素之间的键,也就是C-C键的。前四个键能分别表示:
- Sigma-bond dissociation energy
- Pi-bond dissociation energy
- Double pi-bond dissociation energy
- Bond energy
其他C-C键参数可能也有影响,但这四个对我们本例影响是最主要的,因此作为范例,只优化了这4个参数。
因此我们直接执行:
cp ffield ffield_min cp ffield ffield_max
然后分别修改ffield_min和ffield_max里面,这四个参数的值,优化这四个参数的结果,将会在ffield_min和ffield_max设定的下限和上限之间。可以酌情分别在原值基础上减小、增大约30%。
生成开关文件ffield_bool
执行命令:
adfreport ffield -ffield-bool > ffield_bool
因为只优化4个参数,所以只将ffield_bool文件中,四个参数对应位置的数字,从0改为1。
5. mcff.run文件
注意根据实际需求,灵活修改(参数的含义在本文前面已经进行了说明):
- mcbeta可以用默认值;
- mcdbet设置的越小越好,降温越慢,蒙特卡洛模拟效果越好,但所需的步数就越多,默认值实际上已经比较小了;
- mcffit原则上,步数越大越好;
- mcmxst可以改到比较大,配合mcffit。
到这里为止,所有文件都齐全了,都放在当前目录里面。
6. 优化力场
在命令行运行:
chmod 700 mcff.run ./mcff.run
生成的summery.txt列出了蒙特卡洛的过程,比较重要的数据是能量误差(Current error这一列数据),当然误差越小越好。生成的ffield_best是优化得到的力场,修改成自己希望的名字,例如CHO_modified.ff即可。
使用该力场,在ReaxFF中优化,我们得到的层间距3.41埃,很接近DFT的结果3.355埃:

注意,这里我们没有优化二面角参数,也没有在trainset.in里面限定二面角,所以为了保证层间距,优化键能的同时,二面角的结果变差了,石墨烯变得不是平面的了。