
这是本文档旧的修订版!
电场下打开硅烯和双层石墨烯的带隙
版本:2017.dev
在本教程中,您将学习如何在ATK批量计算中插入金属栅电极并使用它们施加电场。您将使用这些功能来研究在电场存在下双层石墨烯和硅烯中带隙的打开。假定您已经熟悉了VNL的基本功能。

双层石墨烯
构建一个双层石墨烯结构
1.创建一个新的空项目,打开Builder(按钮![]() )。
)。
2.从Database中导入石墨(不是石墨烯!)。
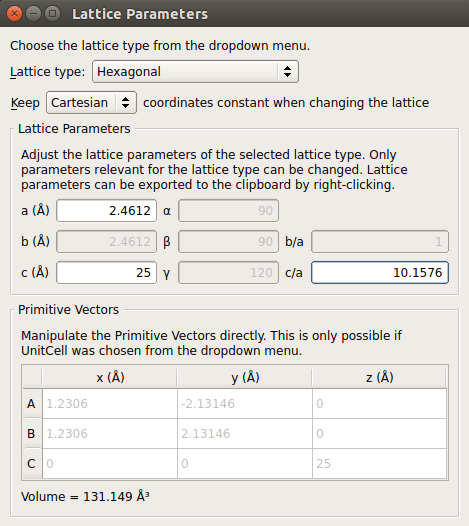
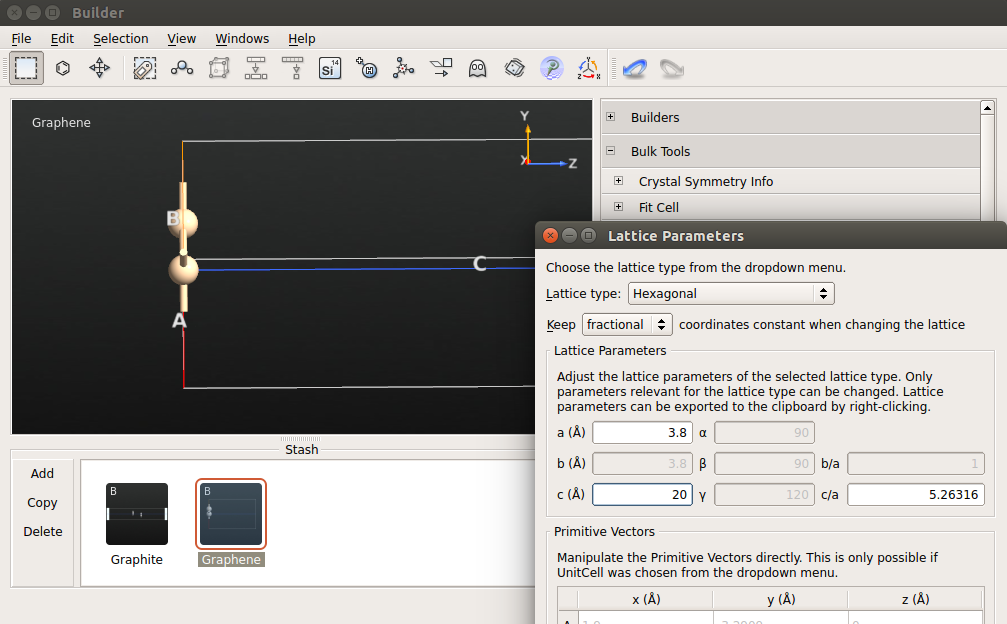
3.打开Bulk Tools Lattice Parameters。选择笛卡尔坐标系,设置晶格常数c为25 Å。
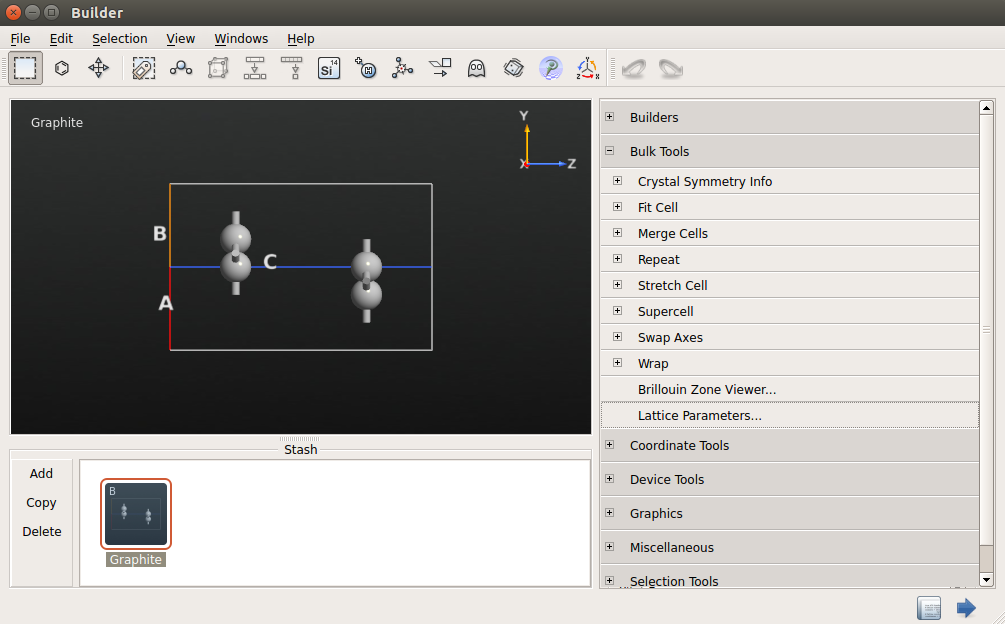
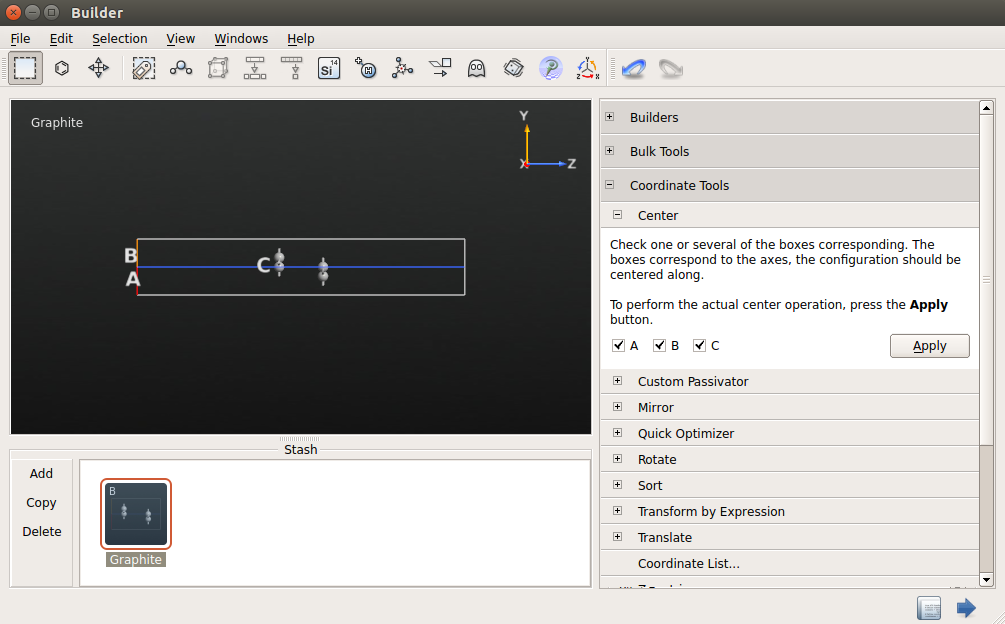
4.打开Coordinate Tools Center,点击“Apply”使体系居中。
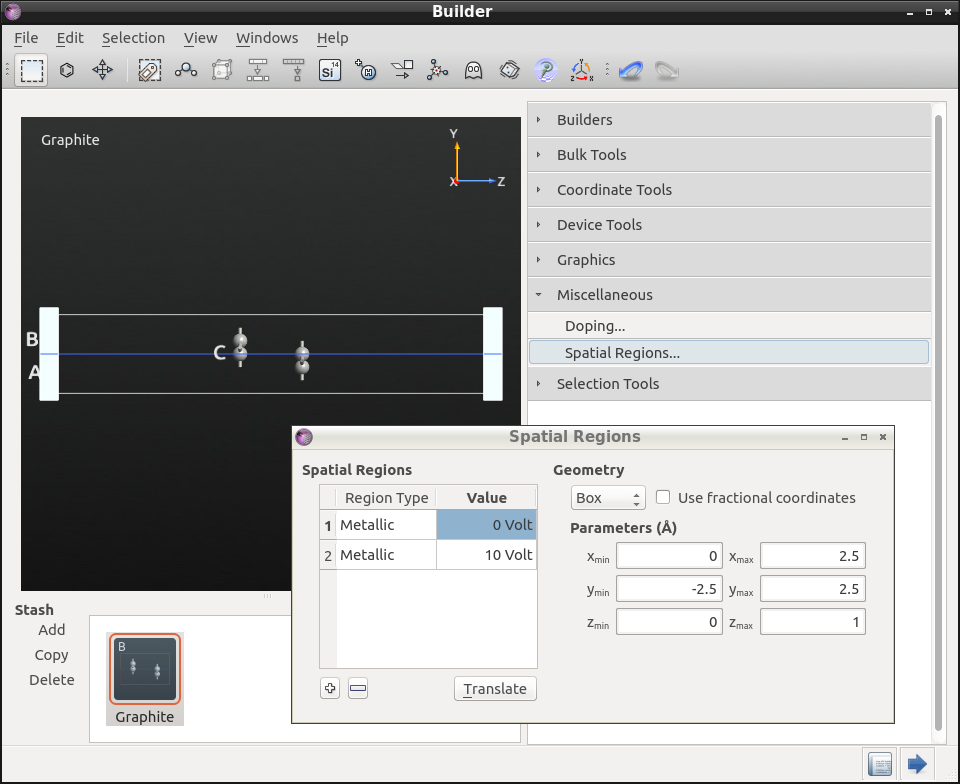
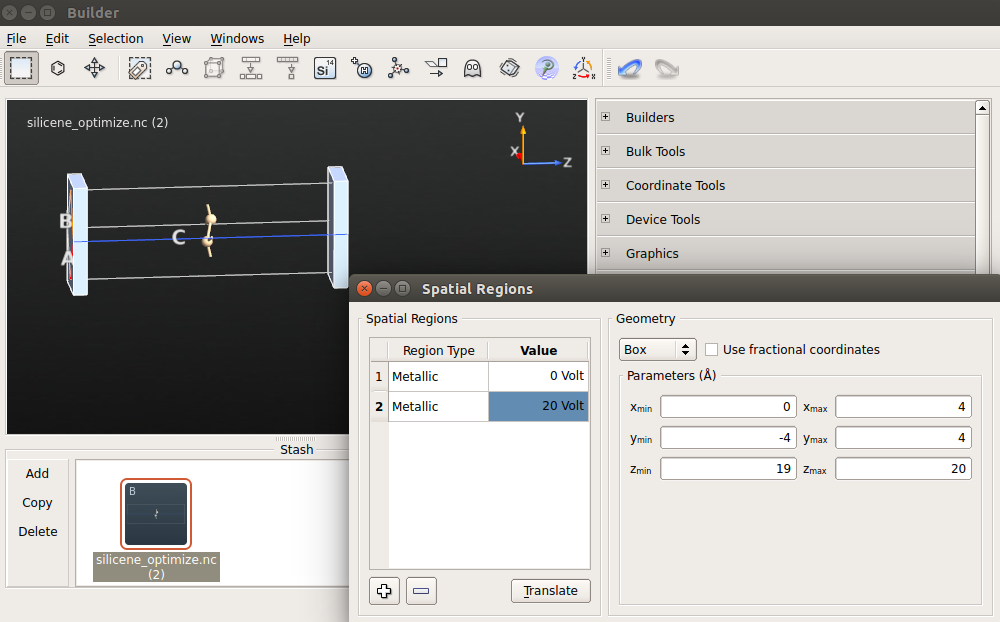
5.为插入金属电极,要先打开Miscellaneous Spatial ![]() Regions…,右键单击表格(表头为Region Type/Value的下方区域),然后单击“Insert region”两次。
Regions…,右键单击表格(表头为Region Type/Value的下方区域),然后单击“Insert region”两次。
6.设置第一个金属区域的电压为0 V,第二个为10 V。改变它们的大小是为了使它们覆盖整个六方晶胞(参见下图)。如果这个区域超出了晶胞之外也没关系,这些部分在计算中会被忽略掉。
施加的电压在z方向上为4 V/nm,略微超过了在实验中发现的能够打开带隙的电场。
计算和分析
然后我们将分析静电场下双层石墨烯的能带结构。
1.发送双层石墨烯结构到Script Generator。
2.插入一个New Calculator模块。
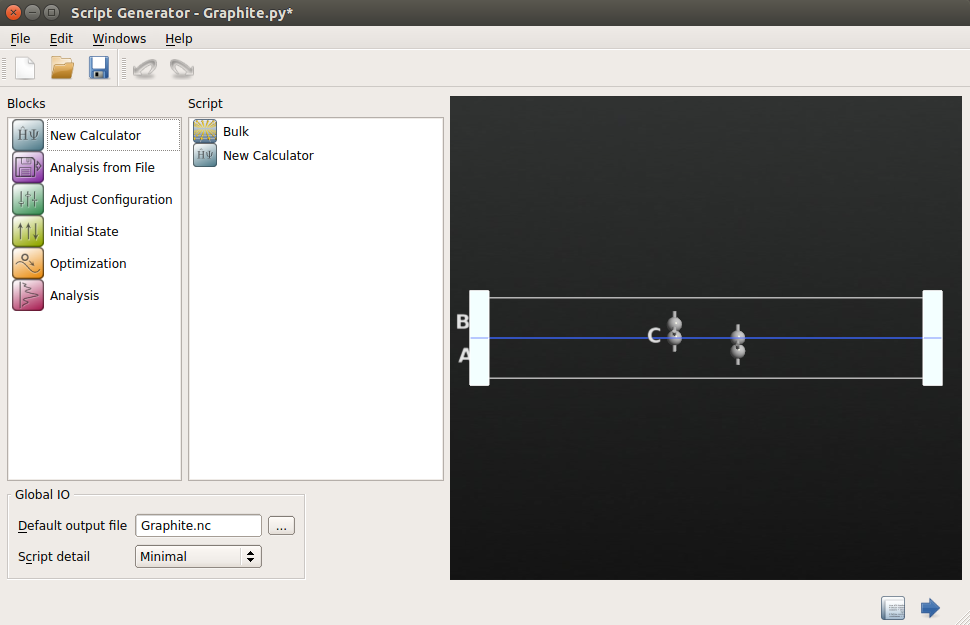
3.打开New Calculator模块,选择“ATK-SE: Extended Hückel”方法。设置k点为9×9×1以确保较高精确度。
4.在“Huckel basis set”下选择可以很好地描述sp2键合的“Cerda. Carbon [graphite]”基组。
5.取消勾选“No SCF iteration”选项执行自洽计算。
警告
包含自洽计算是极其重要的部分,否则施加的场将完全不会产生任何影响。
6.点击“Poisson solver”,在Solver下方选“Multi-grid”,Boundary Conditions中C方向两侧选“Dirichlet”。

注意
对于这个特定的计算,您可以在C方向上选择任何边界条件,因为金属栅极无论如何都会固定边界的电位,但当边界处需要一个恒定的电位值时,“Dirichlet”是在物理上的正确条件。
7.点击Analysis![]() Bandstructure,添加Bandstructure模块。
Bandstructure,添加Bandstructure模块。

8.双击Bandstructure模块,更改Brillouin zone route为G, M, K, G。将每个分割点之间的间隔点设置为200。

9.命名输出文件为“bilayer_graphene_efield.nc”。
10.将计算发送到Job Manager,将出现在窗口中的Python脚本保存并运行计算。运行时间不会超过20-30秒。
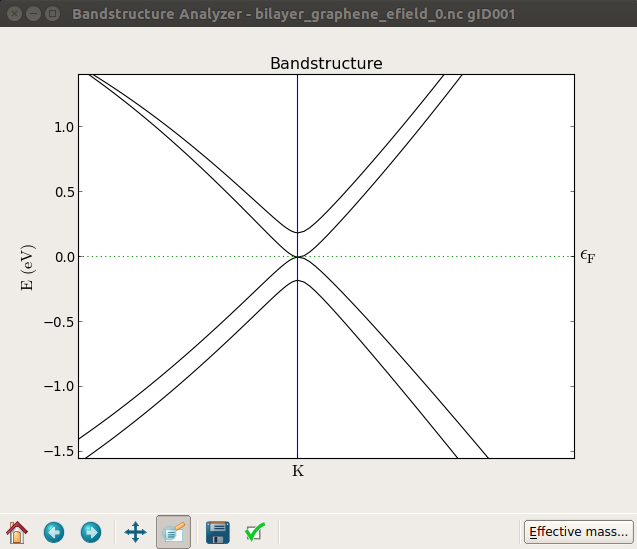
计算完成后,将能带结构可视化,放大K点附近的图像,随着电场强度的增大,带隙逐渐变大。下面的图从上到下分别显示施加电场为0 V、10 V和20 V双层石墨烯的能带结构。

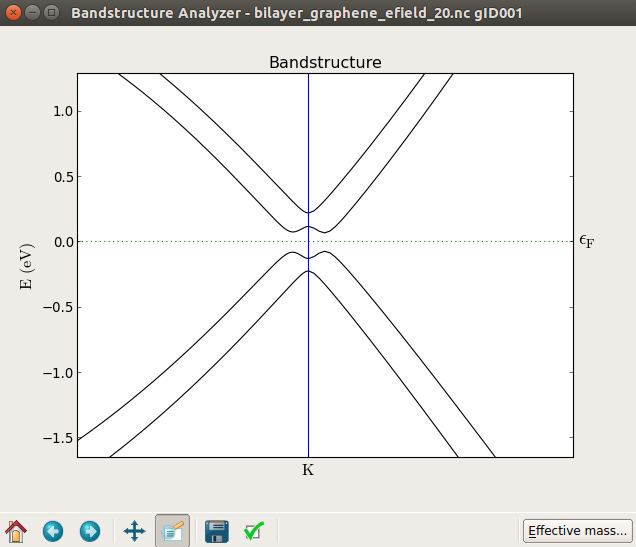
注意
- 您也可以使用DFT,结果相似。
- 计算中没有关注层间距的问题,您可以尝试稍微改变一下,然后观察能带结构的变化。但是请注意,不能采用DFT/GGA优化层间距,因为色散校正的缺失使优化找不到最低能量状态(各层受范德华力的约束,在标准GGA中没有考虑到)。由于误差消除,使用LDA偶尔可以,但结果不可靠。
硅烯
要研究电场对硅烯的影响,首先您需要一个很好的模型。该方法将从石墨烯开始将其转化为硅烯,优化几何结构,然后重复实施与计算电场下双层石墨烯相同的步骤。

几何结构优化
1.在Builder里,实施与上述对双层石墨烯相同的操作步骤,从Database添加石墨烯(注意这次不是石墨)到Stash。
2.选中2个C原子(同时按住鼠标左键和键盘上的Ctrl键),使用“Periodic Table”工具里的 ![]() 将其替换为Si原子。
将其替换为Si原子。
3.硅烯的晶格常数与石墨烯的并不完全相同,但无需猜测其值,几何结构优化就可以确定。为使晶格参数的优化从一个合理的猜测开始,单击Bulk Tools Lattice Parameters设置参数。确定保持分数坐标不变,将晶格常数a设为3.8 Å,c增大到20 Å。
4.点击Coordinate Tools ![]() Center,使结构居中。
Center,使结构居中。
5.当两个Si原子在同一平面时的结构能量是势能面中的局部最小值。因此,点击几次“Rattle”工具 ![]() 给坐标增加一些小的扰动,这样做可以确保结构优化收敛到全局最小值。
给坐标增加一些小的扰动,这样做可以确保结构优化收敛到全局最小值。
6.按下“Send To”按钮 ![]() 把结构发送到Script Generator。
把结构发送到Script Generator。
7.在Script Generator里,双击模块栏的下列按钮插入New Calculator和OptimizeGeometry模块:
 New Calculator。
New Calculator。 Optimization OptimizeGeometry。
Optimization OptimizeGeometry。
8.k点设为21×21×1可保证较高的精确度,交换关联函数选择GGA-PBE。原则上您可以使用默认的DoubleZetaPolarized基组,但此处选择“Tight Tier 1”会得到较好点的结果。

9.在OptimizeGeometry模块,设置一个较小的force tolerance(0.01 eV/Å)和更小的stress tolerance(0.0005 eV/Å3)。在晶胞的x和y方向上不做约束。

10.命名输出文件为“silicene_optimize.nc”,保存Python脚本。
11.将脚本发送到Job Manager,保存脚本并运行作业,计算大约需要10分钟。将ID为“gID001”的构形放到Builder,观察其晶体结构,您会看到一个优化得相当好的硅烯片,屈曲约0.5 Å,晶格常数优化为3.86 Å。这两个值与文献报道的结果均一致[1-2]。
能带结构
使用优化过硅烯的结构,重复上述将双层石墨烯插入金属栅极并在电场下计算能带结构的步骤。请注意,在硅烯的案例中,您需要确保在XY平面上的空间区域更大,因为晶胞也更大了。采用20 V作为第二个电极上的电压。
注意
您应该采用和结构优化时相同的计算器设置,计算时长少于1分钟。
从下图可以看出,零场(上部)处的能带结构和加了20 V电压的栅极(底部)之间产生了约0.1 eV的带隙。尝试施加不同的电压值,您会发现如参考文献[3]中所示:带隙基本上随电场增大呈线性增加。
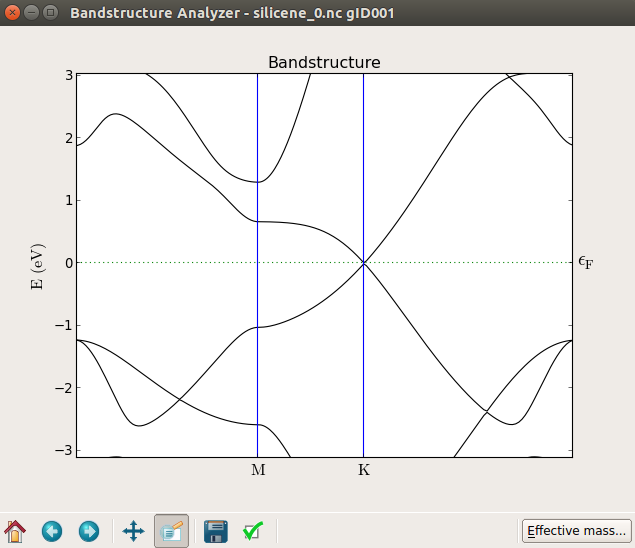
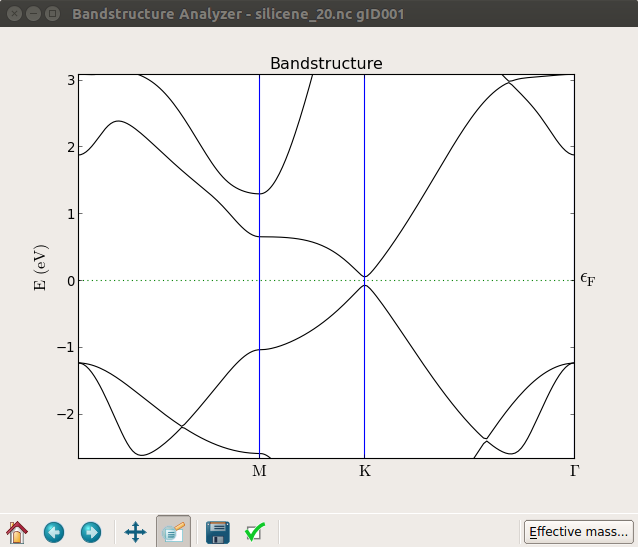
请注意,如果您仔细看会发现,在本例中带隙并不完全精确的在K点。
注意
如果将横场施加到单个石墨烯层上,则不会出现间隙。这是因为石墨烯是完全平坦的,场只能使势能做固定的移动。在硅烯的例子中出现间隙是因为薄片会自然地弯曲。
参考文献
补充参考
一个关于六方BN的相似研究,可参见