
器件体系的建模与计算
简介
本文以氢分子放置在锂的单原子线中间的模型为例,介绍QuantumATK最重要的功能–电子输运计算。本例中的模型非常简单,因此计算可以在很短的时间里完成。然而本例中包括的物理概念和效应对很多实际体系都是适用的。

计算采用密度泛函理论(DFT)方法与非平衡态格林函数(NEGF)方法结合。更多关于计算的理论细节可以参考QuantumATK的手册。
进一步了解如何进行完整、可靠的器件计算,请参考:使用QuantumATK研究电子输运。
提示
本教程使用特定版本的QuantumATK创建,因此涉及的截图和脚本参数可能与您实际使用的版本略有区别,请在学习时务必注意。
用于输运计算的体系几何结构
器件体系的基本结构
用于器件计算的结构模型与普通的周期体系或非周期体系都不一样,而是两者的组合:两个周期体系部分分别代表左右电极(或者是晶体管的源、漏电极),中间的非周期部分是决定器件功能的关键部分,可以是一个分子、石墨烯、纳米管、多层结构等各种体系,这部分对于电子和空穴的透射起到了主要的散射作用。
除了源、漏电极以外,QuantumATK还支持增加门电极,可以模拟晶体管,具体设置方法可以参考相关的实例教程。
QuantumATK中的器件模型由三个相邻的区域构成:左电极、右电极以及位于中间的中央区域(Central Region)。(如下图)

由于电极部分是由周期性体系构成了,而中间区域是有有限的非周期体系构成的,因此它们连接的部分需要一个过渡区域。这部分是左右电极自然延伸而成的表面(Surfaces),分别与左右电极类似。但表面通常会有重构(原子排列不同于块体电极部分)。
此外,为计算的稳定性考虑,QuantumATK要求中央区域的两端最少有一个周期的电极体系延伸部分(electrode extensions),这部分原子排布与电极始终保持完全一致。
电极的延伸部分和表面部分构成了屏蔽区域(Screening Region),这部分存在的意义是在自洽计算的过程中让中间的“分子”结构平滑的过渡到两端的块体结构。
- 这里提到的QuantumATK处理输运的方法是弹性弹道散射,散射来自于表面和分子部分,但是不包括声子的散射。
- 中央区域和电极对输运都一样的重要,如果某一部分是绝缘体或半导体,零偏压下就不会有电流。
从中央区域构建器件
上文解释了QuantumATK中的电极模型实际上在中央区域中已经定义了(电极延伸部分),这也是构建双电极体系的最便捷方法:先构建包含电极延伸部分的中央区域,再扩展周期电极部分。
构造中央区域
本例中使用的模型是氢分子作为杂质镶嵌在锂原子线中间,如下图中用不同颜色条标出了不同的区域。

本文最后一部分介绍如何正确构建中央区域模型,这里先直接给出模型的脚本:
- central_region.py
# Set up lattice vector_a = [6.0, 0.0, 0.0]*Angstrom vector_b = [0.0, 6.0, 0.0]*Angstrom vector_c = [0.0, 0.0, 39.3702095157]*Angstrom lattice = UnitCell(vector_a, vector_b, vector_c) # Define elements elements = [Lithium, Lithium, Lithium, Lithium, Lithium, Lithium, Hydrogen, Hydrogen, Lithium, Lithium, Lithium, Lithium, Lithium, Lithium] # Define coordinates fractional_coordinates = [[ 0.5 , 0.5 , 0.03955015], [ 0.5 , 0.5 , 0.11865069], [ 0.5 , 0.5 , 0.19775123], [ 0.5 , 0.5 , 0.27624721], [ 0.5 , 0.5 , 0.35642874], [ 0.5 , 0.5 , 0.43295913], [ 0.5 , 0.5 , 0.48952536], [ 0.5 , 0.5 , 0.51047676], [ 0.5 , 0.5 , 0.56704061], [ 0.5 , 0.5 , 0.64357304], [ 0.5 , 0.5 , 0.72375085], [ 0.5 , 0.5 , 0.80224875], [ 0.5 , 0.5 , 0.88134932], [ 0.5 , 0.5 , 0.96044984]] # Set up configuration bulk_configuration = BulkConfiguration( bravais_lattice=lattice, elements=elements, fractional_coordinates=fractional_coordinates )
保存文件为central_region.py,把文件拖动到Builder上即可导入结构。(也可以把以上脚本文本拷贝到剪贴板,然后在Builder中选择Add→From Clipboard)
添加周期电极部分
在Builder中选择右侧工具栏中Device Tools→Device from Bulk。

这个工具自动探测中央区域的左右部分的周期性,给出添加电极的多种可能性。本例中两端的锂原子间距相同(约3.11Å),周期性明显,当然二倍、三倍、四倍等周期也是存在的。
尽管看上去选择最小的电极周期是可以的,但实际上却行不通。实际体系通常需要大于7-8Å的电极长度,小于这个长度的电极可能会导致计算结果错误。关于这一点的详细解释请参考双电极体系中如何正确设置电极。
本例中应该选择三个锂原子长度(约9.3Å)作为电极。
用左侧工具中的“Split Device”工具可以把器件的电极部分单独拆分开来,用来进行电极部分的能带计算等。
器件结构的其他说明
关于器件还有一些需要注意的地方。
- 输运方向限定为Z方向。因此电极单胞、中间区域的C轴应与Z方向平行。其他两个胞矢A、B应与Z垂直,即位于XY平面内。
- 电极不必是同种材料,但是左电极、右电极、中央区域的A、B胞矢必须一样。
- 屏蔽区域的层数应该进行收敛性测试。
周期性与边界条件
中央区域和电极在QuantumATK中都定义为块体结构(Bulk Configuration),因此是作为三维周期体系来处理的。当把三个部分结合成器件结构(Device Configuration)时,A、B方向仍然是周期结构,但是只有电极部分的C方向是周期结构,实际上是半无限周期结构(Semi-inifinite),从中间向两边无限周期重复。
在脚本里器件结构是这样定义的:
device_configuration = DeviceConfiguration( central_region, [left_electrode, right_electrode] )
需要注意的是,尽管左电极变量名为left_electrode,右电极为right_electrode,但实际上左右电极的变量名是任意的,仅通过先后顺序来定义哪个是左电极。
在输运计算中,中央区域并不是作为周期体系来处理,而是采用开放边界条件,电荷可以流出或流入中央区域。
由于A、B方向限定为周期结构,因此对于一位的纳米线或二维的片层结构,需要在A或B方向增加足够的真空区域,以确保AB方向周期性重复的体系不会相互作用。真空层的厚度可能需要测试一下收敛性。
小结
QuantumATK中的器件结构是描述中央散射区结构耦合在两个半无限电极结构的模型。构建这种结构的最便捷的方法是先构建包含左右电极的中央区域,再转换成器件。详细的建模过程参见本文最后一部分。
用图形界面Script Generator工具设置计算
下面介绍如何设置器件的计算。
在Script Generator中打开器件结构
将上面的器件结构传送到Scripter Generator(直接拖动或者电极右下角Send to),显示如下。

设置计算参数
设置透射谱计算:
- 双击“Blocks”中的New Calculator,将其添加到Script部分;
- 双击Analysis,选择
TransmissionSpectrum; - 将输出文件名改为
li-h2.nc。

进一步修改计算参数,双击“Script”中的New Calculator;为加快计算,将Density mesh cut-off设为50 Hartree。

点击OK关闭对话框。
双击“Script”中的TransmissionSpectrum。将能量范围设置为[-2,5] eV,选择Self-energy calculator为Krylov(最快的方法)。

点击OK。
运行计算脚本
将脚本传送到Job Manager(右下角拖动或点击“Send to”按钮选择Job Manager)

点击Run Queue开始计算。出现的窗口中会显示计算过程的log信息。本例计算只需要几分钟,计算结束时可以保存log文件以便以后参考。

至此,一个完整的输运计算就结束了(零偏压下)。接下来可以进行结果分析。进一步的其他计算流程是类似的。
结果可视化
简单介绍一下如何查看计算结果。 在VNL文件浏览的窗口中,找到并勾选li-h2.nc文件,在中间的LabFloor窗口中就出现了所有可以查看的结果图标。

选中Transmission Spectrum图标,在右侧工具中点击Transmission Analyzer。新版的Transmission Analyzer中可以交互式的研究透射谱、k空间透射通道、本征透射通道等丰富的信息。

深入分析结果
除了在上面的自洽计算脚本中直接添加其他分析计算以外,QuantumATK还可以稍后再进行其他的分析计算。 这样做很多时候可以把耗时的自洽计算与快速的分析计算分开进行,很多时候更方便。因为往往我们需要先了解体系的基本特性,再根据需要决定进行哪些分析计算,而不是盲目的进行很多没有价值的计算。
例如,我们可以先计算一下透射谱,查看透射最强的能量是多少,再分析该能量处的局域态密度。后面的分析计算都不需要再进行自洽,因此是非常快的。
此外,非自洽的分析计算通常不需要很强的电脑,常常可以在台式机或笔记本电脑上完成。而第一步的自洽计算应该在专用的集群服务器上完成。
器件态密度
在之前的Script Generator窗中,删去New Calculator和TransmissionSpectrum(用Delete键)。 双击Analysis from File,双击Analysis选择DeviceDensityOfStates。

用户也可以直接打开一个新的Script Generator窗口,这样窗口的右侧不会显示结构。但这不影响计算,因为体系的信息将从li-h2.nc文件中读取。
Analysis from File功能可以从过去计算过的结果中读取体系的信息来进行分析,双击它,选择过去自洽计算得到的nc文件。
nc文件中可能包括多个体系的信息,用不同的ID标示(从gID000开始编号),为确定从哪个体系信息开始计算,应首先在VNL LabFoor里确定相应的ID。
点击OK关闭。
双击Script中的DeviceDensityOfStates,设置能量范围为[-5,5] Points为201。

本例中将分析计算的结果保存在相同的li-h2.nc文件中。实际上用户可以选择保存在其他文件中。分开保存有助于管理不同的数据,删去并不需要永久保存的结果等。
将脚本传送至Job Manager并进行计算。
在VNL的文件浏览窗口中选中li-h2.nc,在LabFloor中选中Device Density of States,即可用右侧的2D Plot工具打开分析数据。

在同一个窗口中还科技对态密度进行投影,值得注意的是氢原子上的投影态密度与透射谱形状非常相似。
投影(部分)态密度可以通过在右侧显示的结构上直接选择原子来显示,选择多个原子需要按住Ctrl键。选中原子后还可以在上部选择投影在s、p、d、f等角动量上。
分子投影的自洽哈密顿量(MPSH)
本节讲解如何计算氢分子的本征态,即通常所称的分子投影自洽哈密顿量(MPSH)的本征值谱。 回到Script Generator窗口,删去DeviceDensityOfStates对象,从Analysis增加一个MolecularEnergySpectrum和两个Eigenstate对象。
打开MolecularEnergySpectrum,在Projection Selector中勾选Hydrogen。
打开两个Eigenstate,选择投影到Hydrogen,量子数分别设置为0和1。
用Job Manager运行脚本。结果将出现在li-h2.nc。选中 图标,点击右侧2D Plot即可显示能级谱;点击Text Representation即可打印出全部能级的数值。
图标,点击右侧2D Plot即可显示能级谱;点击Text Representation即可打印出全部能级的数值。
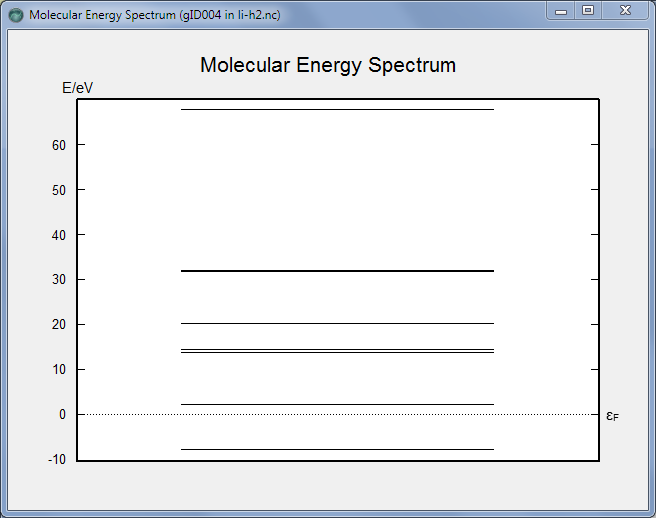
费米能级以下有一个本征态(量子数=0),对应了氢分子的HOMO能级;费米能级以上的第一个本征态(量子数=1)则对应了氢分子的LUMO能级。对比孤立氢分子的能级计算结果,我们发现由于耦合于锂原子中间,氢分子的HOMO-LUMO间距从12.1eV减小到了10.1eV。尤其是LUMO从费米能级以上6eV位置降低到了费米能级以上2eV位置,这解释了为什么氢分子能有比较大的电子透射。
为了作出量子数=0的本征态的图,选中li-h2.nc中的Eigenstate,点击右侧Viewer,在弹出的窗口中选择isosurface或是contour:

- 为了让分子轨道图中显示结构,可以从LabFloor中将结构拖动到轨道图的窗口中;
- 还可以重新拖入分子轨道,选择Contour图,这样就将不同的作图叠加在了一起;
- 使用右侧Property可以任意调整图像的显示效果
本征透射通道
另外一个对我们理解轨道与输运关系非常有帮助的是本征透射通道分析。本征透射通道分析是在给定的能量和k点处,把透射矩阵对角化,得到对该能量、k点处的透射系数的贡献。
在上面的Script Generator中,删去MolecularEnergySpectrum和Eigenstate。从Analysis中增加TransmissionEigenvalues和TransmissionEigenstate。
在默认的参数(能量=费米能级,kx=ky=0)下运行脚本,结果出现在li-h2.nc文件中。
选中TransmissionEigenvalues图标,点击Text Representation,可以看到费米能级处只有一个透射通道(透射系数约为0.7)。
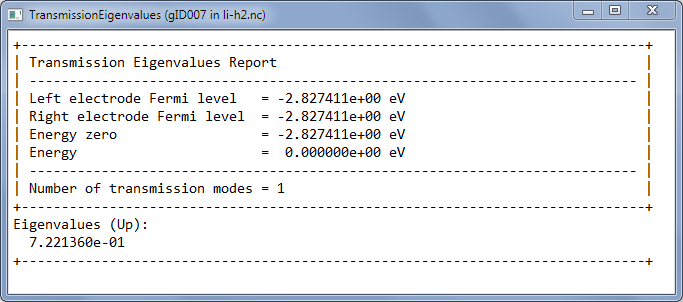
和分子轨道图类似,透射通道也可以可视化。选中透射本征通道,点击右侧的Viewer工具。同样也可以把器件结构拖动到图上以同时显示(颜色显示了相位):

图中可以明显看出,由于中央氢分子的散射作用,右侧的本征态振幅比左侧要小很多。本征函数在左右两侧的波长是一样的,但是经过氢分子时,相位发生了180度翻转。这表明了这个通道的电子透射是通过氢分子的LUMO轨道完成的。
用同一个脚本完成一系列分析计算
多个分析计算可以放在一个脚本中一起完成。例如进行以下计算:
关于器件结构的优化
进行器件计算应该使用合理结构,最“自洽”的方法是用QuantumATK进行结构优化。结构优化时,应该对central region进行优化,为了更快完成,可以尝试:
- 先优化得到块体电极的结构,然后在此基础上创建中间区域;之后冻结中间区域的电极原子(除了电极表面的);
- 给定尽量合理的初始结构(根据直觉考虑,建模时也可以尝试用quick optimizer快速优化);
- 可以先考虑用经验势或半经验进行预优化;
- 仔细优化时可以考虑使用Rigid冻结功能。
当然也可以考虑用VASP优化,VASP优化后直接导入QuantumATK创建器件结构计算输运性质,可以不必再优化。
从2015版起提供了更为合理的(界面)器件优化方案,尤其是当左右电极为两种不同的块体材料时,界面晶胞的优化问题特别重要。详情请参考:如何弛豫器件体系的几何结构。
进一步了解如何进行完整、可靠的器件计算,请参考:使用ATK研究电子输运。