
创建分子-表面体系:苯分子在 Au(111) 表面
版本:2017.0
在本教程中,您将学习如何使用 Virtual NanoLab(VNL) 构建苯分子吸附在金 (111) 表面上两种不同的吸附构型,此处将其表示为 Bz@Au(111)。特别地,您将:
- 使用 Lui 等人在参考文献中 [LRZ+13] 报道过的晶格常数,创建一个 Au 块体。
- 参照 [LRZ+13] 中的参数构建 Au(111) 面的平板模型。
- 在 Au(111) 面的平板模型上附着上苯分子。
- 使用 VNL Builder 中的高级构建工具修改苯分子的原子坐标以构建 Bz@Au(111) 构形。
提示
本教程使用特定版本的QuantumATK创建,因此涉及的截图和脚本参数可能与您实际使用的版本略有区别,请在学习时务必注意。
提示
构建完这些几何结构后,您可以在 ATK 中使用其中一种计算方法对它们进行优化,以执行进一步的分析。
注意
本教程中描述的很多VNL功能都相当通用的,除了表面吸附分子的结构外,还可以构建很多复杂构形,在教程 Building a molecular junction中可以看到更多举例。

工作流程概要
以下是构建 Bz@Au(111) 构形所需主要步骤的概括。
- 创建新的 VNL 项目,打开
 Builder。
Builder。 - 从 Builder Database 中导入块体金的原始晶胞。
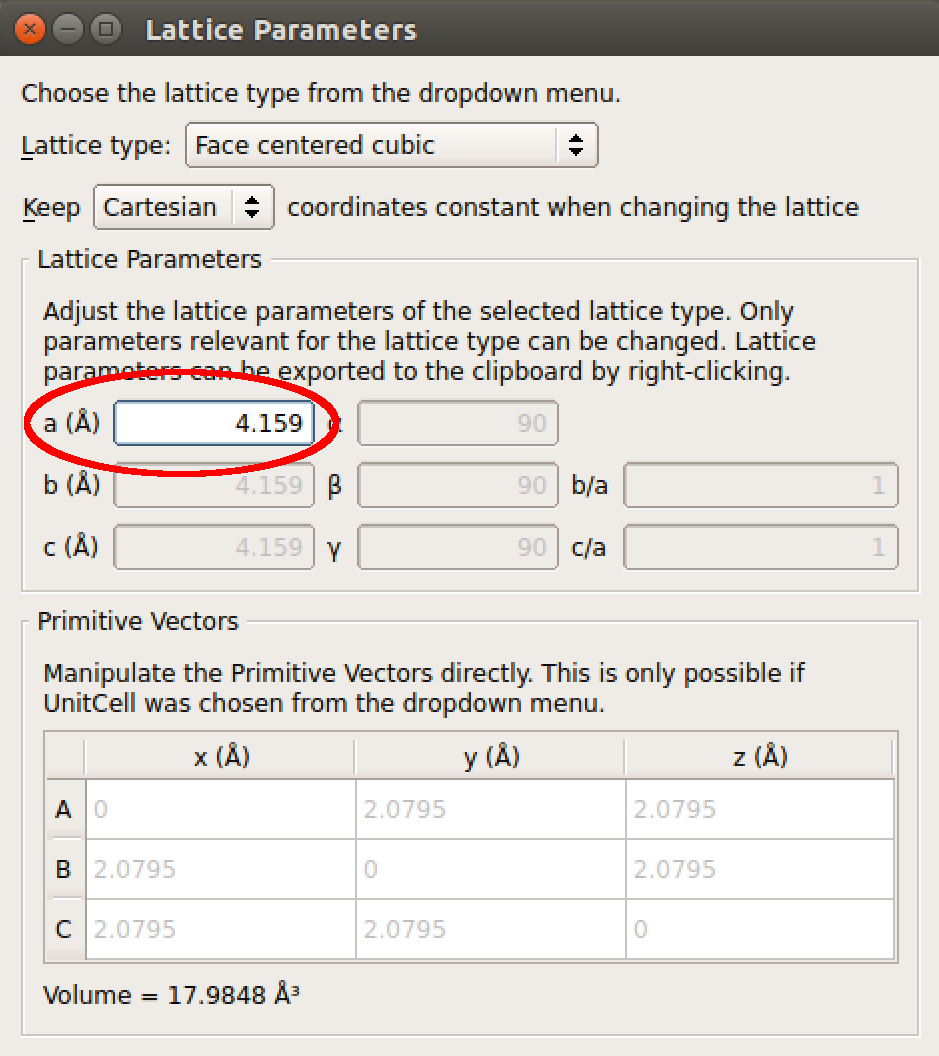
- 参照 [LRZ+13] 的数据,使用 Lattice Parameters 插件修改晶格常数 a 为 4.159 Å,采用 DFT 和 PBE 密度泛函。
- 打开 Surface(Cleave) 插件,构建包含 6 个金属层的 3×3 Au(111) 平板,上方留出 20 Å 的空间。
- 用
 Molecular Builder 工具在平板构形上添加苯分子。
Molecular Builder 工具在平板构形上添加苯分子。 - 选定苯原子,用 Rotate 插件绕y轴旋转分子 90° 使分子可以平行地吸附于平板表面。
- 点击
 图标在分子的几何中心添加一个新原子。
图标在分子的几何中心添加一个新原子。 - 使用
 Move 工具将分子移入 hcp-30° 吸附位。上一步中添加的额外原子用于将分子“吸入”既定位置,随后会将其从构形中删除。
Move 工具将分子移入 hcp-30° 吸附位。上一步中添加的额外原子用于将分子“吸入”既定位置,随后会将其从构形中删除。 - 给构形重命名,并在 Stash 中创建它的副本。
- 给副本重命名,并使用 Rotate 插件绕z轴旋转苯 30°,创建 hcp-0° 吸附构形。
详细说明
构建 Au(111) 表面
您将在此处由金的原始晶胞开始创建 Au(111) 的平板模型。
金块体
打开 VNL,创建新项目。设置标题(此处为 “Bz_Au111” ),选择存放的文件夹路径(脚本、数据文件等),点击 OK 完成创建项目。
点击 OPEN 打开项目,开启您的 VNL 练习。

在 VNL 的主窗口,打开 ![]() Builder,点击 Stash 旁边的 Add
Builder,点击 Stash 旁边的 Add ![]() From Database,打开常用材料的实验结构数据库。
From Database,打开常用材料的实验结构数据库。
在搜索栏搜索 “gold”,选中结果后点击 ![]() 按钮,将结构导入 Stash。
按钮,将结构导入 Stash。
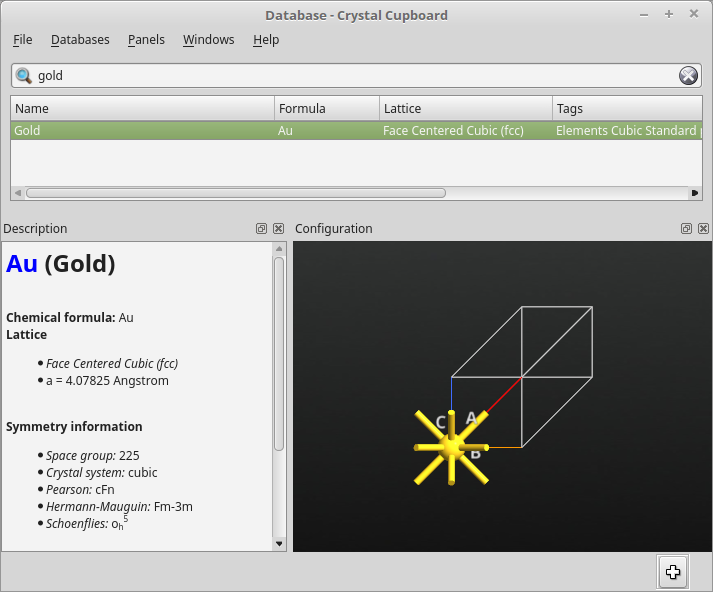
金的原始晶胞已经添加在 Stash 区域,现在可以用 Builder plugins 修改参数。
本例中,依次选择 Bulk Tools ![]() Lattice Parameters 插件,参照文献 [LRZ+13] 中采用 DFT 和 PBE 泛函情况下,将晶格常数 a 设为 4.159 Å。
Lattice Parameters 插件,参照文献 [LRZ+13] 中采用 DFT 和 PBE 泛函情况下,将晶格常数 a 设为 4.159 Å。
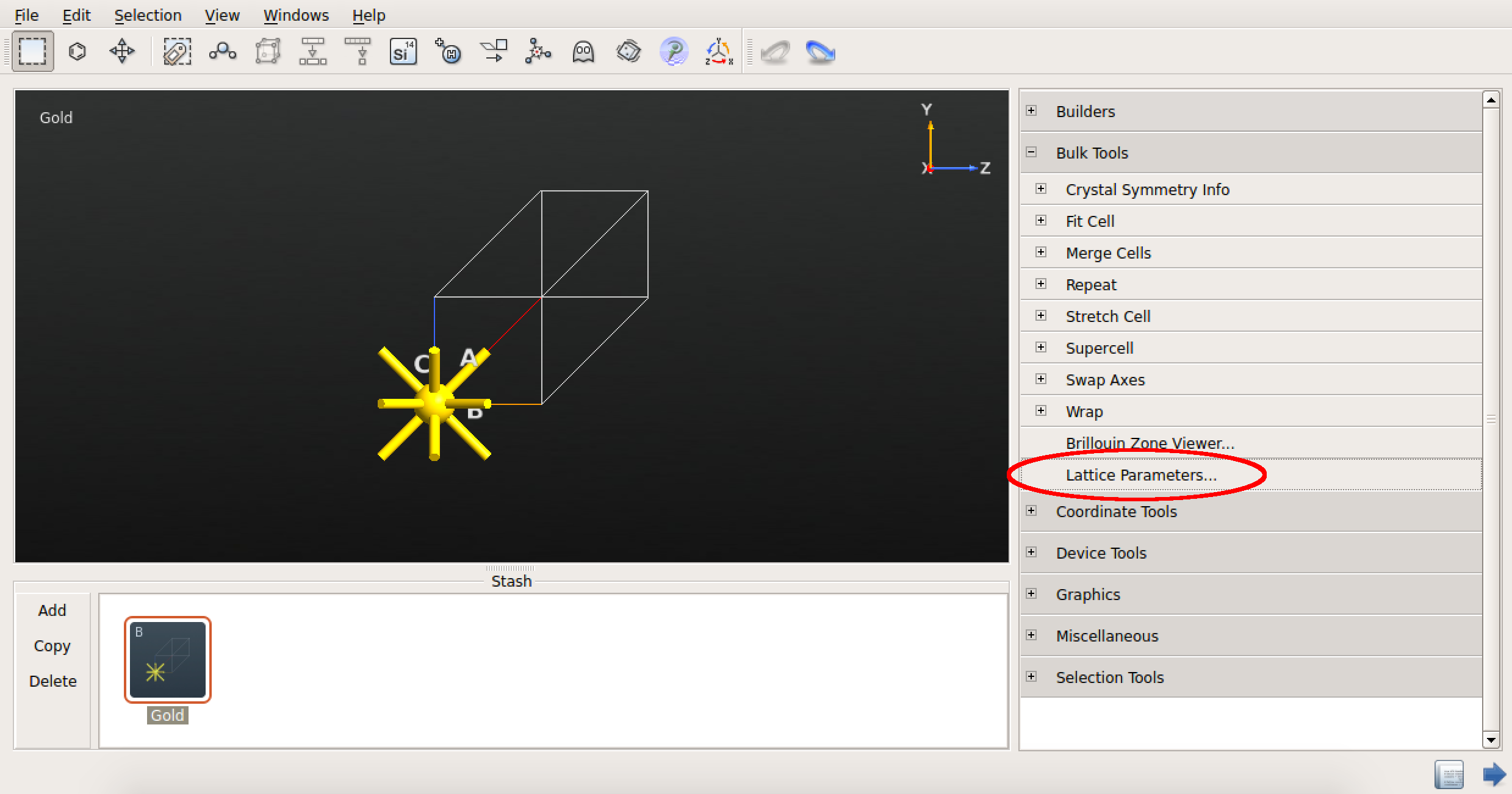
完成以上操作后,关闭插件小工具,返回到 Builder。
Au(111) 平板
下一步是金块体沿 (111) 方向切开,得到 Au(111) 表面的平板模型。
在 ![]() Builder 里, Builders Surface(Cleave) 插件切开金晶体,密勒指数 $(h,k,l)=(1,1,1)$:
Builder 里, Builders Surface(Cleave) 插件切开金晶体,密勒指数 $(h,k,l)=(1,1,1)$:
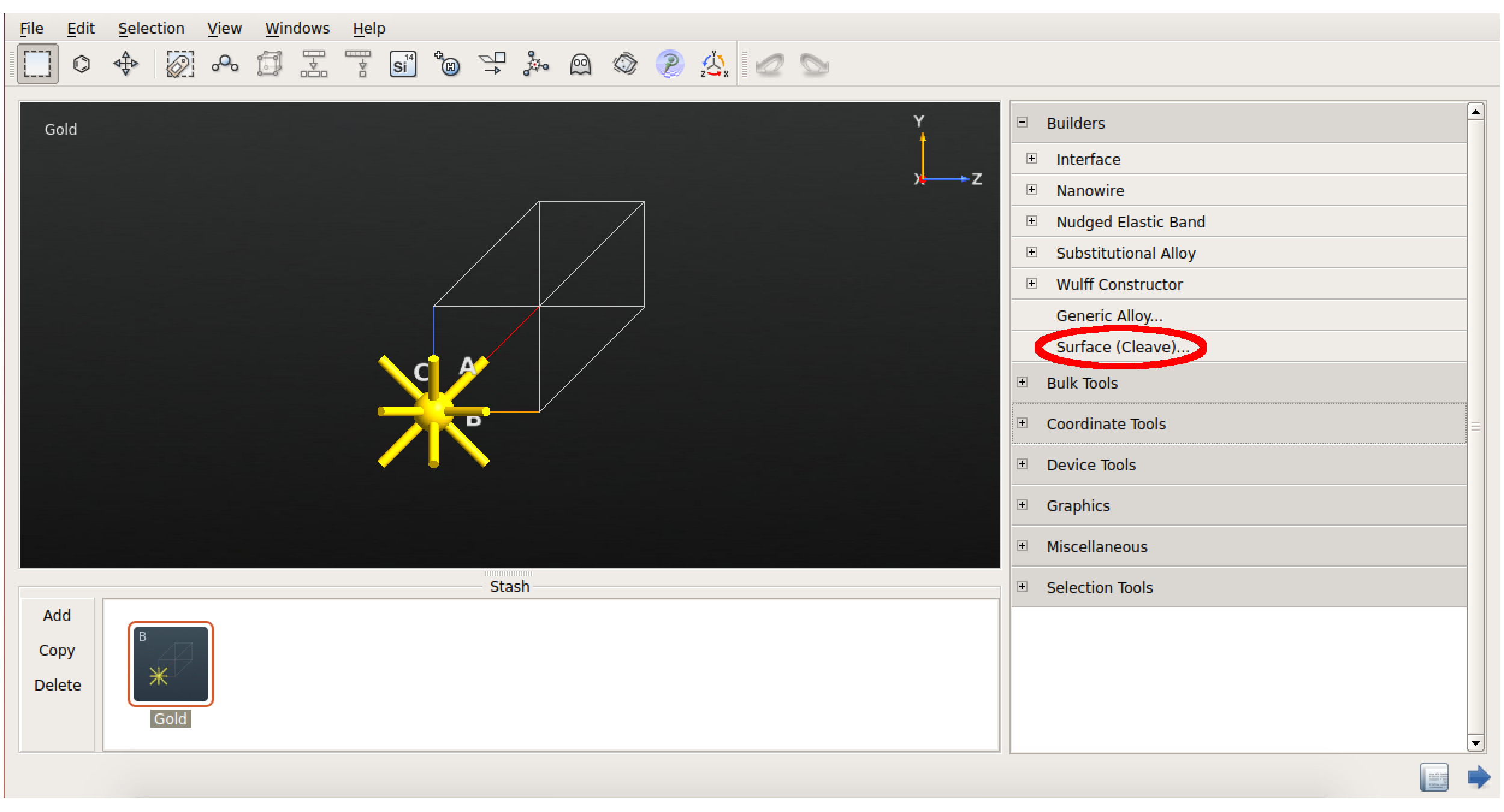
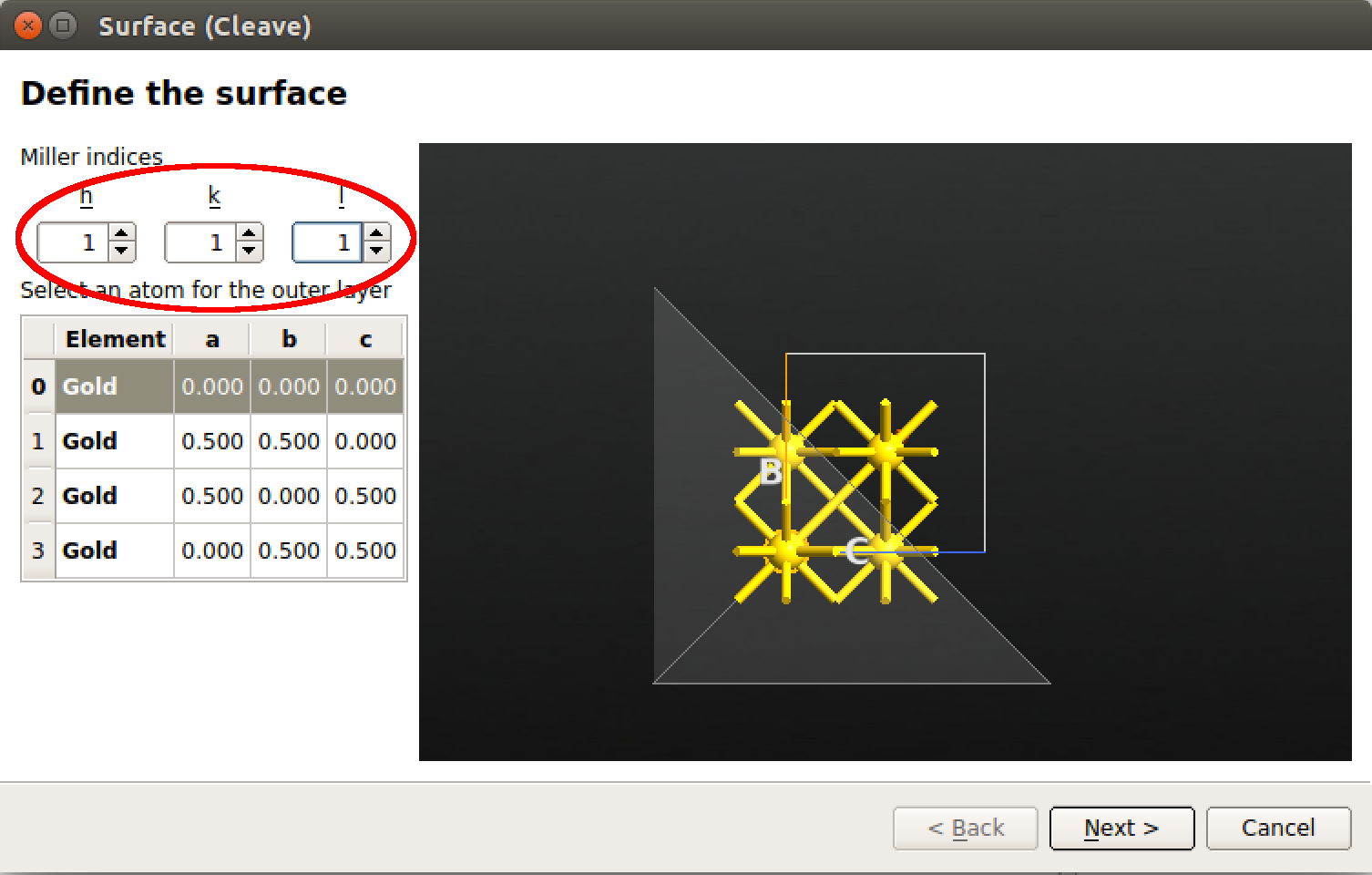
默认的超胞横向大小为 1×1。有些情况下,因为该默认值的存在会非常高效。但在当前的例子中,我们需要 3×3 的平板结构使相邻苯分子的平面间作用力最小化。
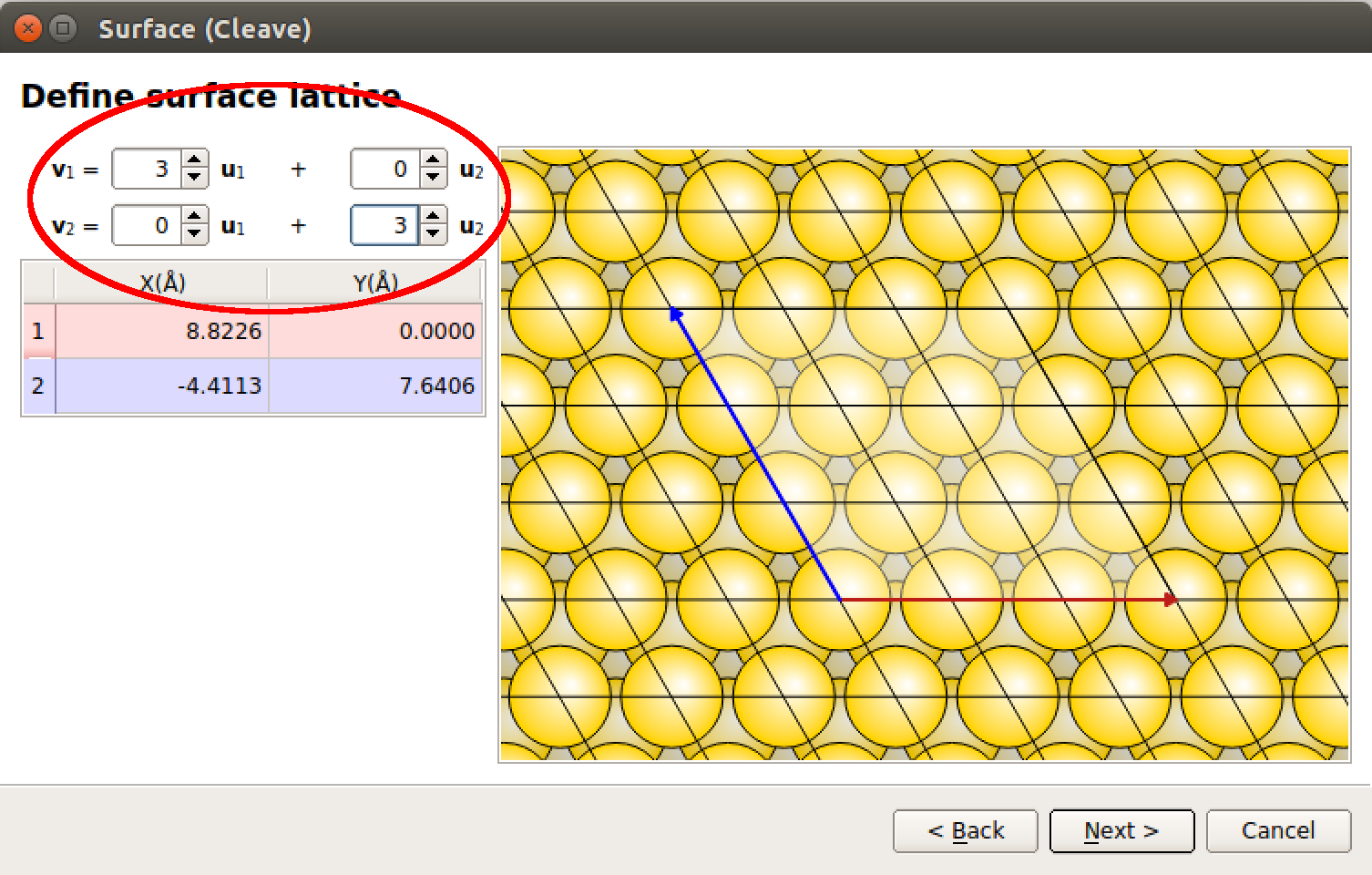
因此,增加晶格矢量 $\mathbf{v}_1$ 和 $\mathbf{v}_2$ 的长度,如此
$$v_1 = 3u_1 + 0u_2$$ $$v_2 = 0u_1 + 3u_2$$
提示
您还可以通过使用鼠标手动地在右边窗口内显示的平面晶胞上移动红色和蓝色矢量实现交互式地修改晶格矢量。
最后,点击 out-of-plane cell vector $\mathbf{v}_3$ is下方的下拉菜单选择 Non-periodic and slab-like 使其形成具有上下方真空的表面。增加 TOP vacuum 值到 20 Å,平板的 Thickness 为 6 层,如下图所示。
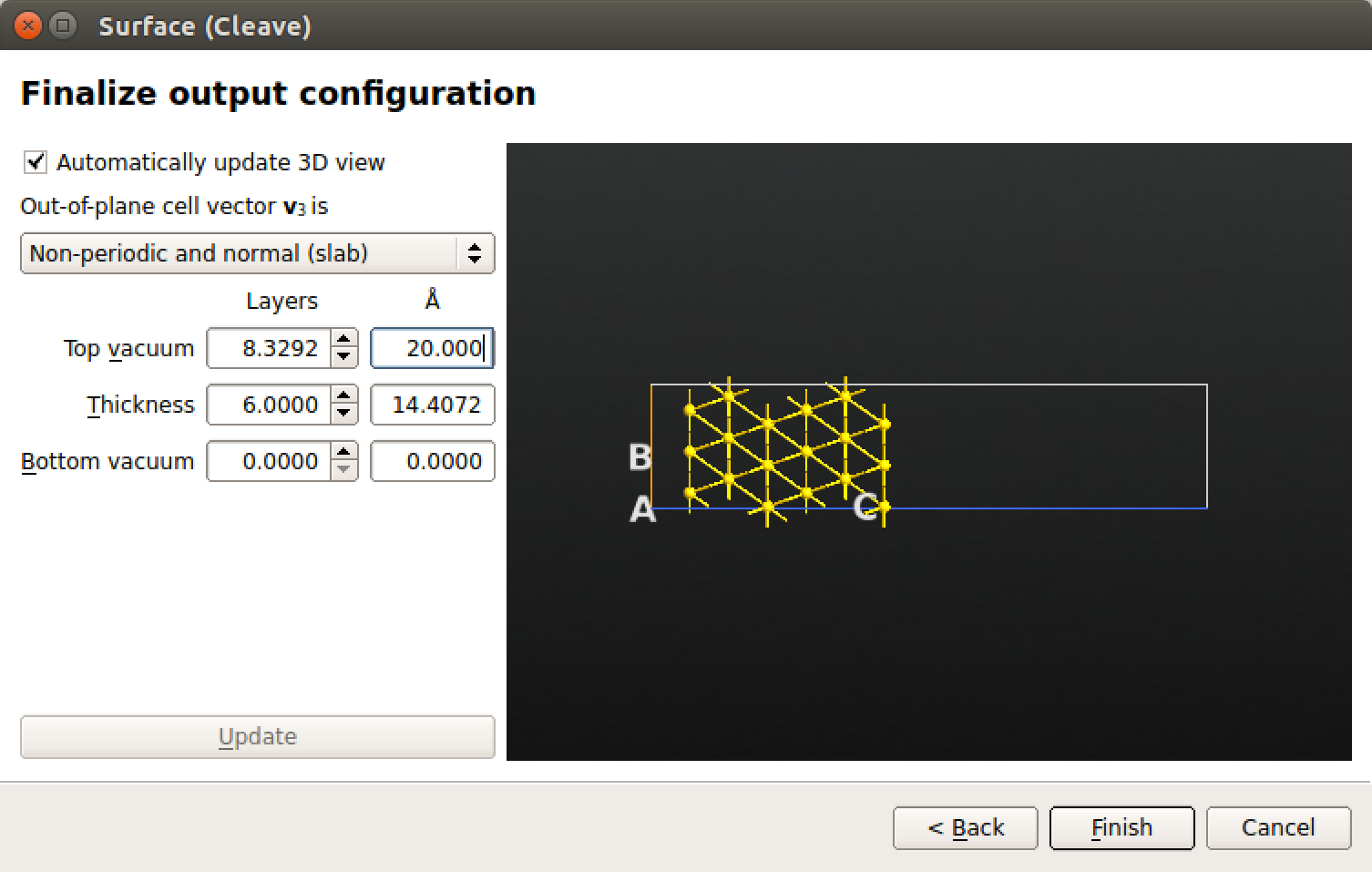
完成以上操作后,点击 Finish。添加 Au(111) 平板构形到 Stash 区,关闭插件小工具。

苯吸附在 Au(111) 面
当然,最后一步就是将苯分子吸附在 Au(111) 平板上。我们的第一个目标就是文献 [LRZ+13] 中报道的 hcp-30° 构形。
选中 Stash 区的 “Gold(111)”,点击鼠标右键或按下键盘上 F2 键将其重命名为 “Au(111)”。复制该结构,再次用鼠标右键重命名为 “hcp30”。
在 ![]() Builder 里,点击 按钮打开 Molecular Builder。
Builder 里,点击 按钮打开 Molecular Builder。
在 Molecular Builder 里,从下拉菜单中点击 Fragments ![]() Simple rings
Simple rings ![]() Benzene。
Benzene。
您现在可以将苯分子插入 Au(111) 构形中:点击 Au(111) 真空区域的某处将分子放置在那里。然后关闭 Molecular Builder 窗口。
警告
需要注意的是,只能点击一次添加苯分子到 Au(111)。因为每点击一次,就有一个分子被添加到同一个位置,新的原子会与那些已经存在的分子重叠!
谨记,您可以随时检查是否有重叠原子:点击 Selection Tools ![]() Close Neighbors 插件,选择一个相对较小的阈值(例如,0.001 Å)。
Close Neighbors 插件,选择一个相对较小的阈值(例如,0.001 Å)。
然后,您需要将分子旋转 90°。首先用鼠标在分子周围画一个矩形选中所有苯原子。打开 Coordinate Tools ![]() Rotate 插件,将旋转轴设为 “y”,旋转角度为 90°,并确保勾选了 Rotate around selection center 的选项框。点击 Apply 执行旋转操作。
Rotate 插件,将旋转轴设为 “y”,旋转角度为 90°,并确保勾选了 Rotate around selection center 的选项框。点击 Apply 执行旋转操作。


下一步,移动苯分子到吸附位置 hcp-30°。首先,确保已选中所有的苯原子,然后点击 ![]() 图标在分子的几何中心添加一个原子。您将利用这个原子为锚点将分子“吸入”表面的上方。
图标在分子的几何中心添加一个原子。您将利用这个原子为锚点将分子“吸入”表面的上方。
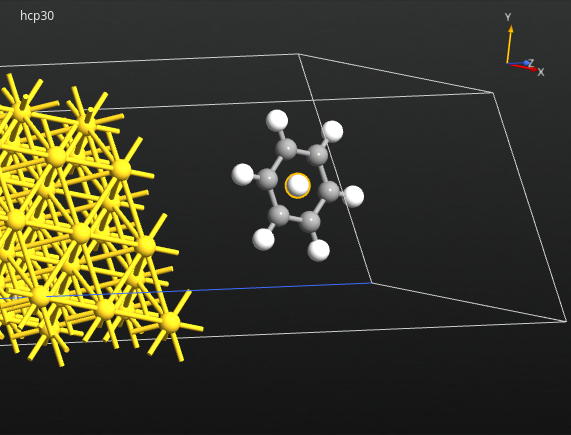
再次全部选中包括额外原子在内的所有苯原子,旋转视图为构形的俯视图。
提示
![]() Camera 工具会非常有用:选 XY 视图平面以获得沿 z 轴的完美视图。
Camera 工具会非常有用:选 XY 视图平面以获得沿 z 轴的完美视图。
点击 ![]() 图标打开 Move 工具,然后点击苯分子中间的额外“吸入原子”,选中其为锚点原子,目标原子周围的阴影应该会变为红色。
图标打开 Move 工具,然后点击苯分子中间的额外“吸入原子”,选中其为锚点原子,目标原子周围的阴影应该会变为红色。

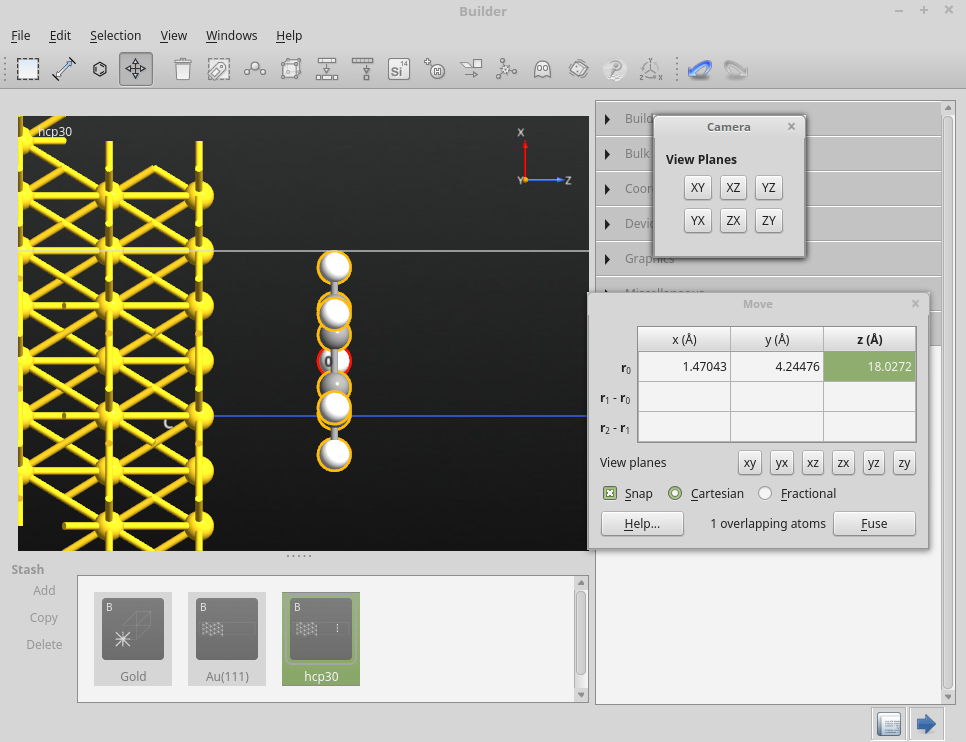
在 Move 面板,确保您勾选了 Snap 的选项框。用鼠标移动锚点原子以拖拽分子将其放在第二个平板层的金原子上。

最后,参照文献 [LRZ+13] 调整分子-表面间的距离为 3.62 Å:在 Move 面板,如下图所示在锚点原子位置 $r_0$ 的 z 坐标处输入 18.0272 Å。
注意
一般情况下,为了设置所需的分子-表面距离,您一定要先知道金最顶层的笛卡尔坐标 Z。这可以通过把指针悬停在 Au(111) 最顶层的一个原子上方而轻易获得。
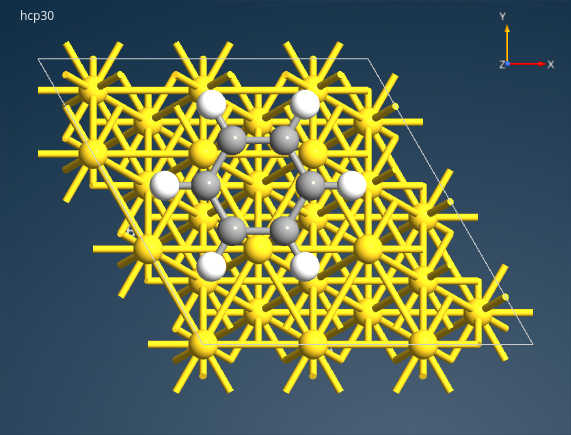
最后,选中锚点原子并删除。至此,您的 “hcp30” 构形应如下图:
提示
您可以把构形保存为 ATKPython 脚本,或者将其导出为一些主流的文件格式。
创建 hcp-0° 构形
hcp-0° 吸附构形与 hcp-30° 吸附构形相关,只要简单的将后者的吸附的苯分子旋转 30° (假设Bz-Au(111)的距离恒定)就可以得到前者。
首先,在 Stash 区域复制 “hcp30”,重命名为 “hcp0”。然后选中新创建副本中的所有苯原子,再次打开 Coordinate Tools ![]() Rotate。
Rotate。
将构形沿表面所在平面的法线z轴旋转 30°, “hcp0” 构形应如下图:

参考
- [LRZ+13] | (1, 2, 3, 4, 5, 6) Wei Liu, Victor G Ruiz, Guo-Xu Zhang, Biswajit Santra, Xinguo Ren, Matthias Scheffler, and Alexandre Tkatchenko. Structure and energetics of benzene adsorbed on transition-metal surfaces: density-functional theory with van der Waals interactions including collective substrate response. New Journal of Physics, 15(5):53046, 2013. URL: http://stacks.iop.org/1367-2630/15/i=5/a=053046.