
这是本文档旧的修订版!
如何进行单重激发态的结构优化(非相对论)
如果我们需要计算荧光或者磷光,激发态的结构优化就是必须要做的事情。如果计算吸收光谱就不需要这一步。一般而言,激发态的几何结构和基态的几何结构不一定具有相同的对称性。因此分子处于激发态的时候,其对称性我们是不清楚的。因此,对于激发态的优化,我们应该将分子的对称性取消掉,然后进行优化。优化之后,如果保持了原有的对称性结构,那就可以认为激发态对称性与基态一样(这种情况其实很少见);如果优化后,对称性消失了,或者降低了,那就只能认为激发态的对称性消失或降低了(大部分情况是如此);也有对称性变高的情况,但非常少。
ADF计算的精确度很高,因此能够让结构收敛到正确的对称性上面去。例如CO2分子,我们从一个对称性最低的结构,例如Cs群的结构去优化,最后收敛后,ADF也能得到D∞h的直线型结构。
由于我们取消了对称性,因此激发态的不可约表示就变成一个A了。最低激发态,也就变成1A了。
参数设置
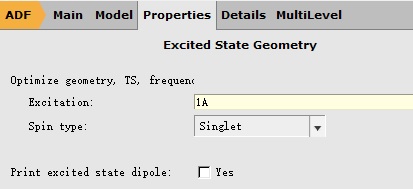
我们如下设置激发态优化的参数(分子结构沿用上一步的结构):

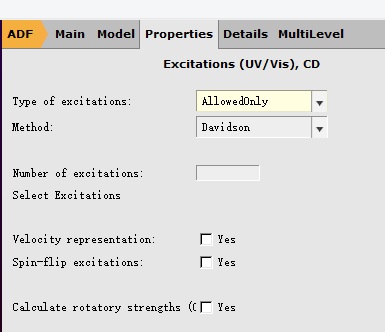

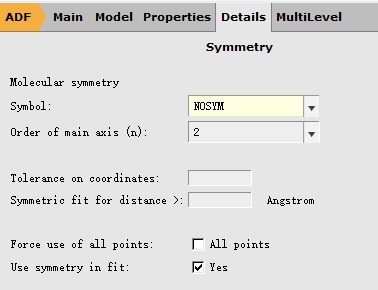
注意,这里1 A表示优化第一个激发态,也就是S1;如果要优化Sn,那么这里就要填写n A。 取消对称性如下:在Details下拉菜单中选择Symmetry得到如下菜单:
保存任务,例如名为:04Excitation_GO,则自动生成04Excitation_GO.adf、04Excitation_GO.run文件。同时会生成04Excitation_GO.pid文件,这个文件对我们一般没有用处。然后执行任务。
结果分析
优化后的分子结构,可以从SCM LOGO > movie中播放的优化过程动画的最后一帧看到。导出该结构可以用如下方式:

也可以选择Save Geometry将其保存为xyz格式。
而优化结束之后的激发态,与前面一样,可以在SCM LOGO > Spectra中查看。可以看到新的S1态的激发能。这其实就对应着荧光的发射峰。组分等也可以同样查看到。在新版中logfile会列出 current energy 实际上就是该激发态的能量,也就是当前结构的基态能量+激发能。
验证激发态优化结果:激发态频率计算,看是否存在虚频
本步计算的分子结构是上一步计算得到的结构。可以从movie中File-update Geometry In Input得到,也可以从File-Save Geometry得到的xyz文件导入。
参数设置(这一步需要非常小心,否则会变成基态的频率计算),参数设置如下:
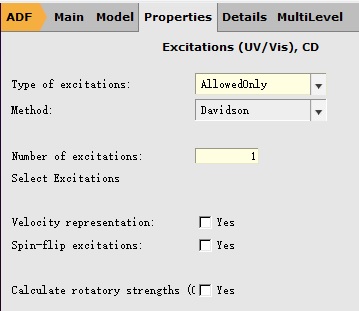
保存任务,例如名为05Freq_of_Excitation。运行计算。
注意:激发态频率的计算,实际上是使用数值拟合的方式去计算。也即是说,需要将每个原子逐个从平衡位置做微小偏移动,然后计算这种微小形变之后的激发能,因此对于像这样的大分子,计算激发态频率将是非常耗时的。如果不考虑对称性的话,计算的结构数大约为原子个数的6倍——也即是说需要计算这么多次激发态。ADF激发态的并行效率很高。因此高核数并行对于这样的计算很重要,否则几乎不可能完成这样的计算任务。总体而言ADF的效率不错,本计算在4核台式机上完成,大约用了4天。如果在16核服务器,基本上1天就足够了。
结果查看
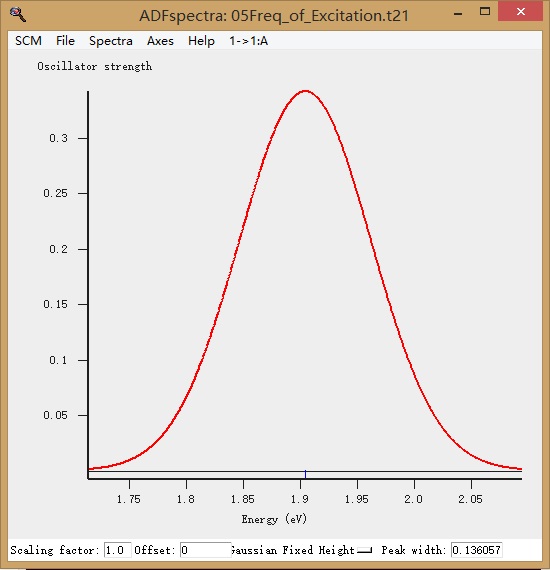
同样地,在SCM LOGO > Spectra中可以查看到振动谱:
首先显示的是激发态(我们上面只计算了1A这一个激发态,因此只有一个峰):
选择振动谱:
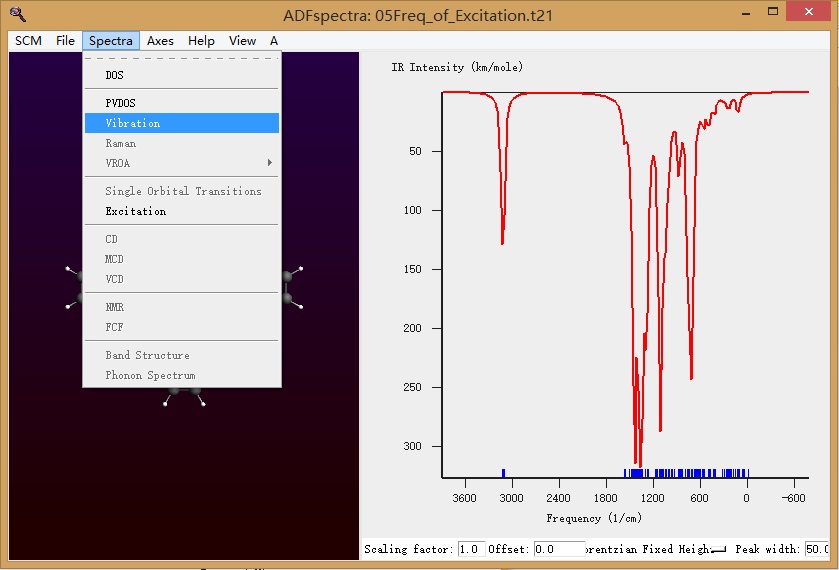
即得到右方的振动谱。振动谱中没有虚频(负数的频率)。