用 DFTB-NEB 研究氨分子翻转反应势垒
在本教程,您将学习利用 NEB 方法计算氨分子翻转反应路径。为加快计算速度,我们将采用基于紧束缚密度泛函(DFTB)方法(dftb.org)。
设置 NEB 对象
- 打开
 Builder,点击 Add
Builder,点击 Add  From Database。
From Database。 - Database 工具打开后,从菜单中选择 Datebases Molecules。在搜索栏输入 “Ammonia”,您将看到如下:
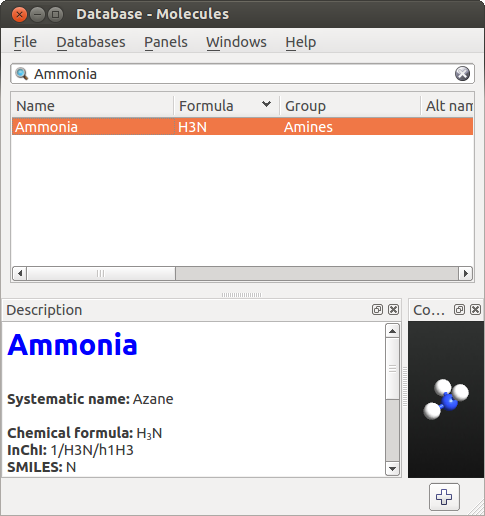
双击搜索结果栏的 “Ammonia”(或者点击 ![]() 按钮),氨分子被添加到 Stash。
按钮),氨分子被添加到 Stash。
注意
运行 NEB 计算时,限制一些自由度是非常重要的。此处主要考虑到的是在氨反应过程中氮原子穿过氢平面移动。因此,定义初始反应路径时,固定氢原子平面是个非常好的做法。这点可以轻易实现,如下所示。
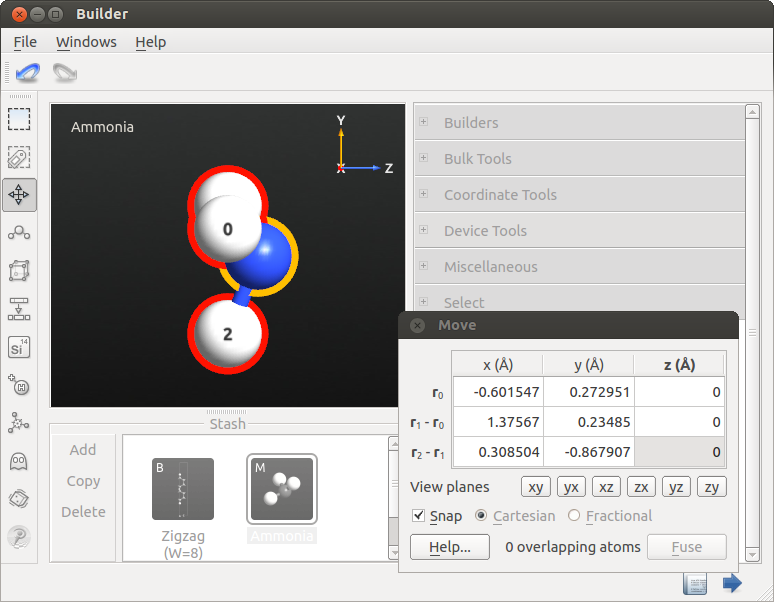
- 在 Builder,激活 Move Tool
 。选中每一个氢原子作为锚原子(标记为红色光环);这就是您要操作的原子。因为 Move Tool 打开时没有一个原子被选中,所有的原子将被同时选中(标记为黄色光环);这些是会被 Move Tool 中的操作影响到的原子。
。选中每一个氢原子作为锚原子(标记为红色光环);这就是您要操作的原子。因为 Move Tool 打开时没有一个原子被选中,所有的原子将被同时选中(标记为黄色光环);这些是会被 Move Tool 中的操作影响到的原子。 - 在 Move 程序窗口,原子 z 坐标处输入值 0,按下 Enter 键。
- 关闭窗口或点击 Select 工具以关闭 Move 工具。
- 在 Stash 区域确保左键单击选中氨分子。然后点击 Copy 按钮,就在 Stash 区添加了另外一个相同的氨分子。
- 打开右侧工具栏 Coordinate Tools 里的 Mirror 工具,保证镜像平面的法线平行于 Z 轴且过原点(这点是默认的),按下 Apply 翻转分子。
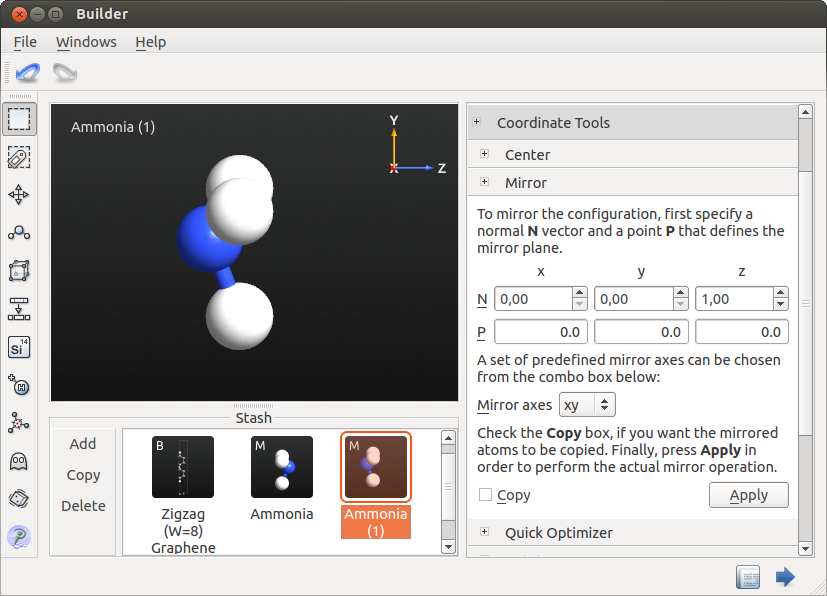
- 您现在已经定义了反应路径的初始状态和最终状态。下一步是通过在这些状态之间插入许多图像来设置 NEB 构形。
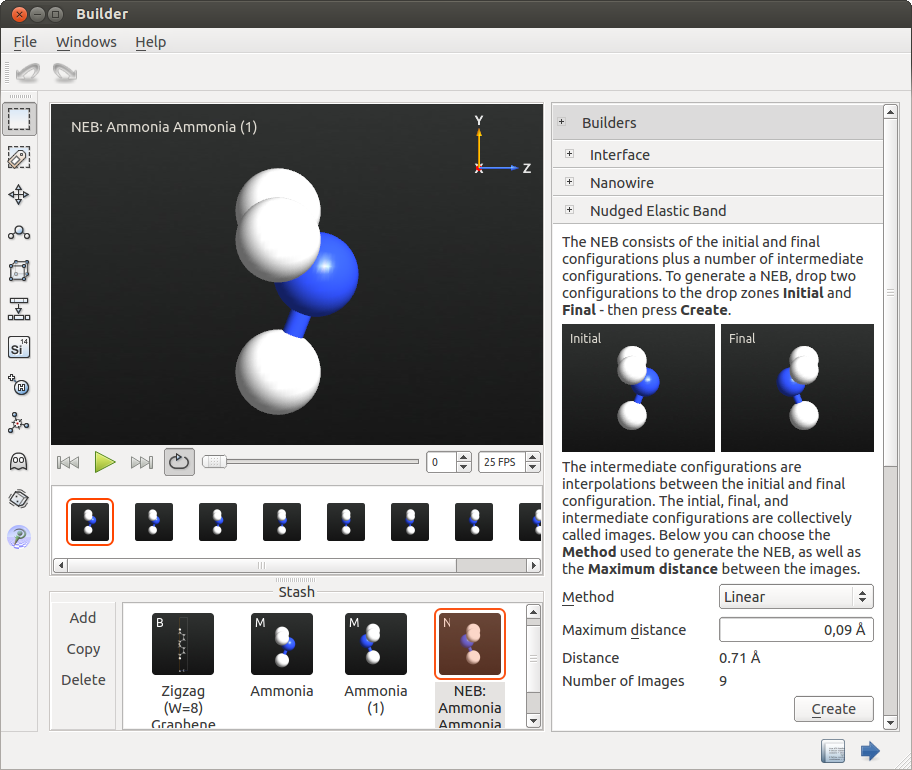
- 为了实现这个目的,请从插件面板中打开 Builders 下的 Nudged Elastic Band 工具,然后将这两个构形拖入 Initial 和 Final 槽位。
- 将图像之间的最大距离设置为 0.09 Å 以获得奇数个图像。这点是非常重要的,因为反应路径是对称的。选择 Linear 方法并按下 Create。现在已经创建了 NEB 对象并添加到 Stash 中。
- 按下 Play 按钮查看优化反应路径时作为起始的不同构形。
运行 NEB 模拟
设置实际运算,采用 Send to 按钮 ![]() 将 NEB 对象发送到
将 NEB 对象发送到 ![]() Script Generator。
Script Generator。
在 Script Generator,添加以下模块:
 New Calculator
New Calculator OptimizeGeometry
OptimizeGeometry
修改默认输出文件名称为 nh3_neb.nc。
打开 ![]() New Calculator,按照如下编辑:
New Calculator,按照如下编辑:
- 选择 ATK-SE: Slater-Koster 计算器。
- 在 Slater-Koster basis set 里,确认勾选了 DFTB [MIO] 基组。
- 不勾选 No SCF iteration 选项框。

打开 ![]() OptimizeGeometry 模块,做如下设置。
OptimizeGeometry 模块,做如下设置。

现在您已经做好了 NEB 优化的准备,发送脚本到 ![]() Job Manager 执行运算。
Job Manager 执行运算。
本次优化依赖于您的电脑将耗时 20-30 分钟。
注意
尽管 DFTB 模型的大多数其他变形都采用逐点静电对势相互作用模型, ATK-SE 则使用泊松解法计算静电相互作用。对于小型体系,例如本例的氨分子,这种方法明显较慢。但是对于大体系,速度很快,采用 $O(N)$ 代替经典的 $O(N^2)$ 按比例缩放。
泊松解法的采用可确保 DFTB 模型与 QuantumATK 的其他计算器相当,因此可用于模拟器件构形且允许使用隐形溶剂模型。
分析 NEB 模拟
- 返回 QuantumATK 的主窗口,选择文件 nh3_neb.nc。
- 选择文件里的最后一个 NEB 构形(ID 号最大的那个)。
- 按下 Show configuration,您将会看到反应路径上计算得到的能量势垒和相对应的构形。
- 通过按下 Play 按钮,反应路径可以展示为动画。
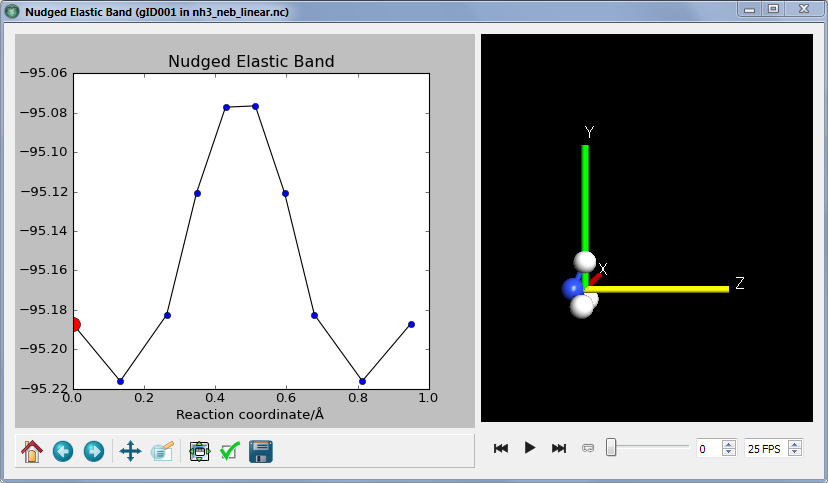
关注
获得的反应势垒并不完全准确,因为终点没有被优化。从第二张图中有能量点比终点还低就能明显看出来。对于使用 NEB 方法的实际研究,应该始终先优化终点。
提示
您可以通过单击反应路径图中的点(并停止动画),仔细检查反应路径中的单个优化构形。
一种快速计算的方法
在上面的计算中,您采用在端点之间线性插值来设置 NEB 路径的初始猜测。然而,在很多情况下,线性路径与优化路径相差甚远。将上述收敛结果的动画与您设置的初始 NEB 路径进行比较,可以看出,当氮原子通过分子中心时,氢原子需要向外移动为氮原子让出空间,然后再移动回来。
如果这种行为能以某种方式成为初始路径的一部分,则可以节省离子动力学中的一些步骤,从而节省计算时间。然而,由于氢原子的初始位置和最终位置是相同的,所以不能通过线性插值捕获。
但是,还有其他方法可以生成初始路径,有些可以在 ATK 中实现。
- 返回到 Builder,选择 Nudged Elastic Band 插件。
- 保持图像距离,但是更改 Method 为 Image Dependent Pair Potential,创建 NEB。
- 如果您现在检查初始路径会发现,它已经包含了氢原子的行为。这是采用 IDPP 方法做 NEB 运算的其中一个优势。
- 按照如上的相同步骤通过
 Scripter 运行 NEB 计算,您会发现这次时间快了 30%,但结果是相同的。
Scripter 运行 NEB 计算,您会发现这次时间快了 30%,但结果是相同的。
注意
IDPP 方法与 Halgren-Lipscomb 方法相似,也可以在 ATK 中实现(但不太适合本例的体系)。
参考
- S.Smidstrup et al., “Improved initial guess for minimum energy path calculations”, J. Chem. Phys. 140, 214106 (2014) link
- T.A. Halgren and W. N. Lipscomb, Chem. Phys. Lett. 49, 225 (1977)