添加、组合、修改经典势函数
简介
新的 ATK-ForceField 软件包提供了许多涵盖广泛元素和系统的预定义经典势。但是,如果您对某个特定的势感兴趣,而其尚未成为预定义集的一部分,只要有支持的基础势,您就可以轻松地添加这些参数并创建自己的势。您还可以修改现有势的单个参数将其调整为特殊的参考系统。此外,您可以结合不同的势来涵盖更广泛的元素。本教程简要概述了 ATK-ForceField 的此功能。
注意
对于 2017 年之前的 QuantumATK 版本,ATK-ForceField 计算器位于 ATK-Classical 之下。
有关 ATK-ForceField 和基础 TremoloX 软件包中可用势的分类和函数的详细概述及文档,请查看 ATK Reference Manual 的 TremoloX section。在那里,您将找到所有功能的定义和相应的参数。
注意
请注意,如果您需要使用新的势,请务必仔细阅读基础出版物,以确保势函数适合您需要的应用。
如果有可能,您应该将一些获得的基准值与经典势作比较以与实验值或适宜的更高水平的计算如结果一致,如 DFT 模拟。尤其适用于不同势的组合,因为它们可能已经在特定上下文中被参数化了,且并不总是能够确保应用在完全不同的环境中。
从零开始添加一个新的经典势函数
在本节中,您将学习如何将取自文献中势函数的一个新参数集添加到 ATK-ForceField 计算器。您将按照指导从头开始完成势函数的设置以解释 TremoloX 势集的各种内容。在实践中,通常从相似的势开始会更合适些,将其发送到 Editor,并将 Script details 设置为 “Show defaults” 用以报告势的所有详细信息,然后就是在 Editor 中编辑参数。
作为第一个例子,您将考虑一种简单的氧化钛晶体。最受欢迎的二氧化钛的经典势之一是由 Matsui 和 Akaogi(MA)[1]提出的,因为它很好地再现了晶体和无定形二氧化钛体系的基本性质。稍后,您将设置一个用于无定形氧化物系统[2]模拟的势。
设置系统
为了设置合适的测试系统,请打开 Builder,单击 Add ![]() From database,搜索 “TiO2”,然后选中并添加 Rutile 晶体结构。
From database,搜索 “TiO2”,然后选中并添加 Rutile 晶体结构。
您现在可以将构型发送到 ScriptGenerator 设置所需的优化、分子动力学或分析脚本。现在,您可以选择任何一个任意计算器作为占位符。一个典型的模拟脚本可以包括 Calculator、Optimization ![]() OptimizeGeometry 和 Analysis
OptimizeGeometry 和 Analysis ![]() ElasticConstants 模块。为获得准确的结果,请将 OptimizeGeometry 设置中的 maximum force 和 maximum stress 值以及 ElasticConstants 设置中的 maximum force 设置为 0.001 eV /Å(eV /Å3)。此外,取消勾选 OptimizeGeoemtry 控件中
ElasticConstants 模块。为获得准确的结果,请将 OptimizeGeometry 设置中的 maximum force 和 maximum stress 值以及 ElasticConstants 设置中的 maximum force 设置为 0.001 eV /Å(eV /Å3)。此外,取消勾选 OptimizeGeoemtry 控件中 Constrain cell 的选框以优化晶格参数。
将脚本发送到 Editor。
设置势函数
在 Editor 中,找到定义势的模块(即 BulkConfiguration 之后和 OptimizeGeometry 模块之前的所有部分)并删除它。
为定义一个新的 TremoloX 势,您首先需要初始化一个新的 TremoloXPotentialSet 数据块。您可以通过添加以下内容实现:
potentialSet = TremoloXPotentialSet('MatsuiAkaogi_TiO')
圆括号中的字符串定义了势的名称,但对其功能没有任何影响。
目前为止,势集还是空的,因此您需要添加所有必需的组件。您应该首先添加 ParticleTypes。在本例中,通过在脚本中插入以下两行来添加一个钛和一个氧粒子类型:
potentialSet.addParticleType(ParticleType(symbol='O', mass=15.9994*atomic_mass_unit, charge=-1.098)) potentialSet.addParticleType(ParticleType(symbol='Ti', mass=47.867*atomic_mass_unit, charge=2.196))
粒子电荷取自文献[1]。MA 势由两种交互类型组成:
1.Tosi-Fumi(通常也称为 Buckginham)型势(即指数、排斥项贡献和有吸引力的范德瓦尔斯部分的组合)。 2.静电库仑相互作用力模拟原子的粒子电离。
第一部分可以使用 TosiFumuPotential 添加,实现了势函数
$$V(r) = A e^{B(\sigma -r)} - \frac{C}{r^6} - \frac{D}{r^8} \, .$$
如果将上述公式与参考文献中给出的等式进行比较[1],您会注意到最后一项不是必需的,这就意味着 D 必须设置为零。注意文献[1]中的能量单元单位为 kJ / mol。参数必须以此为单位或转换为电子伏特。A,B,C,D 和 $\sigma$ 参数可以按照如下设置:
potentialSet.addPotential(TosiFumiPotential(particleType1='O', particleType2='Ti', A=0.811696*kiloJoulePerMol, B=5.154639*Angstrom**-1, sigma=2.8171*Angstrom, C=1215.0*kiloJoulePerMol*Angstrom**6, D=0.0*eV*Angstrom**8, r_i=6.0*Angstrom, r_cut=7.5*Angstrom)) potentialSet.addPotential(TosiFumiPotential(particleType1='O', particleType2='O', A=0.979056*kiloJoulePerMol, B=4.27350*Angstrom**-1, sigma=3.2678*Angstrom, C=2916.0*kiloJoulePerMol*Angstrom**6, D=0.0*eV*Angstrom**8, r_i=6.0*Angstrom, r_cut=7.5*Angstrom)) potentialSet.addPotential(TosiFumiPotential(particleType1='Ti', particleType2='Ti', A=0.644336*kiloJoulePerMol, B=6.49351*Angstrom**-1, sigma=2.3664*Angstrom, C=506.25*kiloJoulePerMol*Angstrom**6, D=0.0*eV*Angstrom**8, r_i=6.0*Angstrom, r_cut=7.5*Angstrom))
每个势函数的前两个参数决定了势的哪两种粒子类型相互作用。附加参数 $r_{i}$ 和 $r_{cut}$ 确定范围和截断行为。在本例中,对于小于 6 Å 的距离,势函数遵循原始形式,而对于较大距离,如高达 7.5 Å 时会调用平滑函数。在较大距离处,假设这些相互作用为零。
通过添加合适的 Coulomb solver 来考虑静电相互作用。所有的 Coulomb solver 都使用您为粒子类型定义的粒子电荷。ATK-ForceField 中的默认算法是 CoulombDSF,采用一个截断和阻尼移位力(DSF)来平滑过渡[2]。为使这种方法获得较高精度(这对于静态计算尤其重要,如弹性常数,声子或晶格常数),必须选择足够高的截断值(例如 ≥ 15 Å)。在这里,我们将有选择地使用 smooth-particle-mesh-Ewald 方法(CoulombSPME),它能够自动处理库仑相互作用的远程贡献。Coulombsolver 可通过以下添加:
potentialSet.setCoulombSolver(CoulombSPME(r_cut=9.0*Angstrom))
最后,您必须初始化实际的 TremoloXCalculator,并将创建的势集合作为参数传递。之后,您必须将计算器附加到块体构型:
calculator = TremoloXCalculator(parameters=potentialSet) bulk_configuration.setCalculator(calculator)
脚本的其余部分可以保留原样。您可以在文件 ↓ TiO2_Matsui.py 中找到示例脚本。
现在,您可以将脚本发送到 JobManager 并启动计算。结果应与文献[1]一致。
无定形氧化物的势函数
在本例中,引入的另一种势被设计用于模拟非晶材料,如钛、硅、铪或氧化钽,以及它们的混合氧化物[3]。
注意
从 ATK-2015 版本开始,这种势已经可以作为预定义势 Trinastic_HfOSiTaTi_2013。
为测试势,您可以使用前一个示例中的金红石构型。该势与 MA 势相似,如上所示,但它另外还包含了一个 MorsePotential。
$$V(r) = E_{ij} \left [ \left(1 - e^{-k_{ij}(r-r_0)} \right)^2 - 1 \right ]$$
实际上,文献[3]中提出的公式省略了方括号中最后一项的贡献。然而,由于该项仅表示能量的恒定偏移,对 MD 模拟中的力并无影响,因此我们将在此处忽略这种差异。尽管通过使用 TosiFumiPotential 原则上可以设置类似于前面示例的势函数项,但我们将采用单独地添加所有项呈现另一种可选方法。
为了获得势,您需要一个取自 BuckinghamPotential 一个离散项的指数排斥贡献,因此您将使用 LennardJonesMNPotential,最后用 MorsePotential。
在 Ti-O 相互作用的例子中展示了势的设置。通过从文献[3]插入相应的 element-pair-specific 势参数,其他元素遵循相同的方案。
从初始化一个新的 TremoloXPotentialSet 开始,添加具有相应粒子电荷的粒子类型:
potential_set = TremoloXPotentialSet('Trinastic_JCP2013') potential_set.addParticleType(ParticleType(symbol='O', mass=15.9994*atomic_mass_unit, charge=-1.2)) potential_set.addParticleType(ParticleType(symbol='Ti', mass=47.867*atomic_mass_unit, charge=2.4))
然后定义一个排除 Buckingham 项:
potential_set.addPotential(BuckinghamPotential(particleType1='O', particleType2='Ti', A=5105.12*eV, rho=0.2253*Angstrom, r_i=6.0*Angstrom, r_cut=7.5*Angstrom))
添加色散项。您将仅需要 Lennard-Jones 势中具有吸引力的部分,即 A 参数必须设置为零:
potential_set.addPotential(LennardJonesMNPotential(particleType1='O', particleType2='Ti', A=0.0*eV*Angstrom**12, B=20.0*eV*Angstrom**6, m=12, n=6, r_cut=10.0*Angstrom))
添加 Morse 势:
potential_set.addPotential(MorsePotential(particleType1='O', particleType2='Ti', E_0=0.3478*eV, k=1.9*Angstrom**-1, r_0=1.96*Angstrom, r_i=6.5*Angstrom, r_cut=7.0*Angstrom))
最后,设置一个 Coulomb solver,如 CoulombSPME 或 CoulombDSF 计算静电相互作用力,并初始化一个具有以下势集的新 TremoloXCalculator:
potential_set.setCoulombSolver(CoulombSPME(r_cut=9.0*Angstrom)) calculator = TremoloXCalculator(parameters=potential_set)
在文献[3]中,提出了一种可供选择的强大参数化,可以通过插入相应的参数替代上文展示的那些来实现。其余元素对之间交互的实现也类似。在文献[3]的参数表中,您会发现对于某些元素组合,Morse 能量参数为零,即意味着这些元素之间不需要 Morse 势。完整的势可以在文件 ↓ script_Trinastic_potential.py.txt 中找到。
Tersoff 和 Lennard-Jones 的组合势函数
在本节中,您将学习如何结合 Tersoff 势和 Lennard-Jones(LJ) 势模拟石墨晶胞中的单个锂原子。这种材料组合特别有趣,因为它提供了一个非常简单的锂离子电池中典型阳极的模型。Tersoff 势和 LJ 势的组合通常用于多组分材料,其中各个组分间相互作用较弱,主要是排斥力和范德华力。在本例的石墨中,组分中的强键合相互作用可以由 Tersoff 势或对于该材料的另一种合适的势描述。
在本例中,您将采用文献[4]中的参数。请注意,它们尚未针对此系统进行过广泛测试。因此,它们只应被视为您自己研究的起点。
设置系统
为设置测试系统,打开 Builder,点击 Add ![]() From database 添加一个石墨晶胞。单击 Bulk tools
From database 添加一个石墨晶胞。单击 Bulk tools ![]() Repeat 扩大晶胞,对单胞进行 4x4x2 的重复。
Repeat 扩大晶胞,对单胞进行 4x4x2 的重复。
如下图所示选中一个包含 3 个原子(一个在第二层,两个在第三层)的三角形。
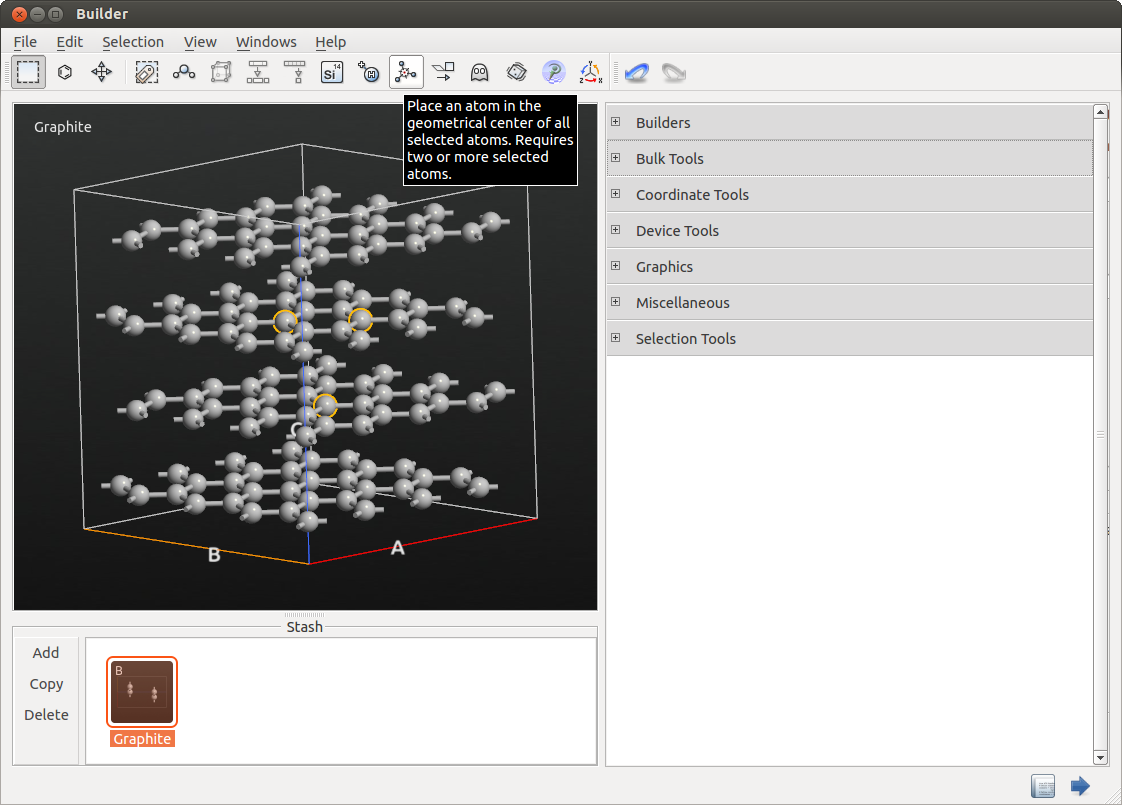
单击工具栏(如图)中的 ![]() 图标,在所选原子的中心放置一个额外的原子。这个原子稍后将被转换为锂,但现在我们将其保留为碳。
图标,在所选原子的中心放置一个额外的原子。这个原子稍后将被转换为锂,但现在我们将其保留为碳。
使用 ![]() 按钮将构型发送到 ScriptGenerator。在 ScriptGenerator 中,添加一个 NewCalculator 对象,然后是您选择的模拟任务,如 Optimize
按钮将构型发送到 ScriptGenerator。在 ScriptGenerator 中,添加一个 NewCalculator 对象,然后是您选择的模拟任务,如 Optimize ![]() MolecularDynamics 模块。打开 NewCalculator 的设置,选择 ATK-ForceField 计算器和 Tersoff_C_2005 势(此为石墨的基本势)。不勾选
MolecularDynamics 模块。打开 NewCalculator 的设置,选择 ATK-ForceField 计算器和 Tersoff_C_2005 势(此为石墨的基本势)。不勾选 Save 和 Print。在 Global IO 的分组框中,将 Script Detail 设置为 “Show defaults”,然后将脚本发送到 Editor。
首先,找到元素列表。将列表中最后一个元素的名称从 Carbon 更改为 Lithium。(如果您已经在 Builder 中完成了这项工作,那么就不可能在 ScriptGenerator 中选择 Tersoff 势,因为它不支持锂。)
如果您现在看一下计算器的部分,将会发现 Tersoff 势的详细定义。这是 Show defaults 选项的缘故。通过 addParticleType() 方法找到添加 “C” 元素定义的行。复制该行(调用两个 addParticleType() ),并按如下所示修改这些行:
potentialSet.addParticleType(ParticleType(symbol='C', mass=12.0107*atomic_mass_unit, charge=None, # Sigma and epsilon from Ref [3]. sigma=3.3611*Ang, epsilon=0.004207*eV)) potentialSet.addParticleType(ParticleType(symbol='Li', mass=6.941*atomic_mass_unit, charge=None, # Sigma and epsilon from Ref [3]. sigma=0.826*Ang, epsilon=0.271115*eV))
对于碳,已添加 LJ 参数 $\epsilon$ 和 $sigma$ 并将其设置为文献[4]中的值。对于锂,首先必须更改符号和原子质量,然后设置相应的 LJ 参数。请注意,本例中我们考虑中性锂原子,即没有定义粒子电荷,因为这是阳极中的优选状态(与阴极中的锂离子相反)。
仅通过指定粒子参数,LJ 势不会变得活跃。您还必须添加相应的势函数。为实现此目的,添加以下两行:
potentialSet.addPotential(LennardJonesPotential(particleType1='C', particleType2='Li', r_cut=9.0*Ang)) potentialSet.addPotential(LennardJonesPotential(particleType1='Li', particleType2='Li', r_cut=9.0*Ang))
这将在碳和锂之间以及锂和锂之间添加 LJ 势。注意,碳原子之间没有 LJ 势,因为这些相互作用由 Tersoff 势充分定义(LJ 只是一个微小的校正)。
在 ATK-ForceField 里的 Lennard-Jones 势的定义为
$$V_{ij}(r) = 4\epsilon_{ij} \left [ \left(\frac{\sigma_{ij}}{r} \right)^{12} - \left(\frac{\sigma_{ij}}{r} \right)^{6} \right]$$
利用以下组合规则获得不同元素间的 $\sigma_{ij}$ 和 $\epsilon_{ij}$ (参见 reference manual)。
您应该始终检查为所选 LJ 参数推荐的组合规则是否与此定义一致。如果没有,您应该使用更为通用的 LennardJonesMNPotential。
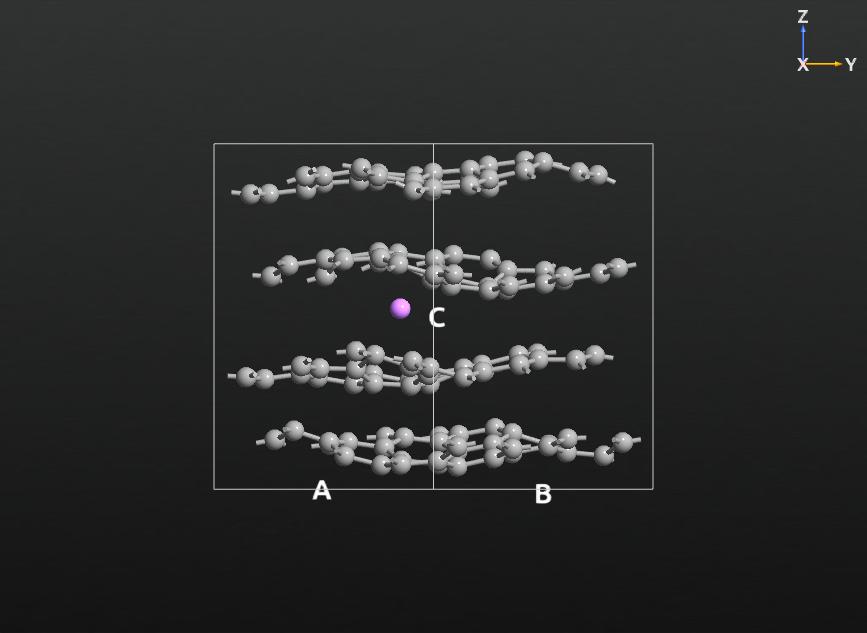
在 ↓ Script_graphite_lithium.py 中可以找到最终脚本的示例。您可以添加与 ATK-ForceField 兼容的任何优化、分子动力学或分析模块。下图显示了锂-石墨系统的 MD 模拟的快照。
MoS2 中的层内和层间内聚力
注意
如果您正在使用 ATK2014,请确保已升级到 ATK2014.3 (或更高版本)以运行本教程的这一部分,因为早期的版本可能会产生一些问题。
在最后一个示例中,您将学习如何使用标记仅在所选原子上定义势。我们考虑一种辉钼矿(MoS2)晶体,如参考文献[4]中所述,它的内部相互作用由 Stillinger-Weber 势描述,而相对较弱的层间相互作用再次由 Lennard-Jones 势模拟,。
为设置系统,请打开 Builder,搜索并添加数据库中的辉钼矿结构。通过在其周围绘制一个矩形来选择一个图层,并添加名为 “layer1” 的标记(通过 Selection Tools ![]() Tags)。对第二层重复以上步骤,命名为 “layer2”。
Tags)。对第二层重复以上步骤,命名为 “layer2”。
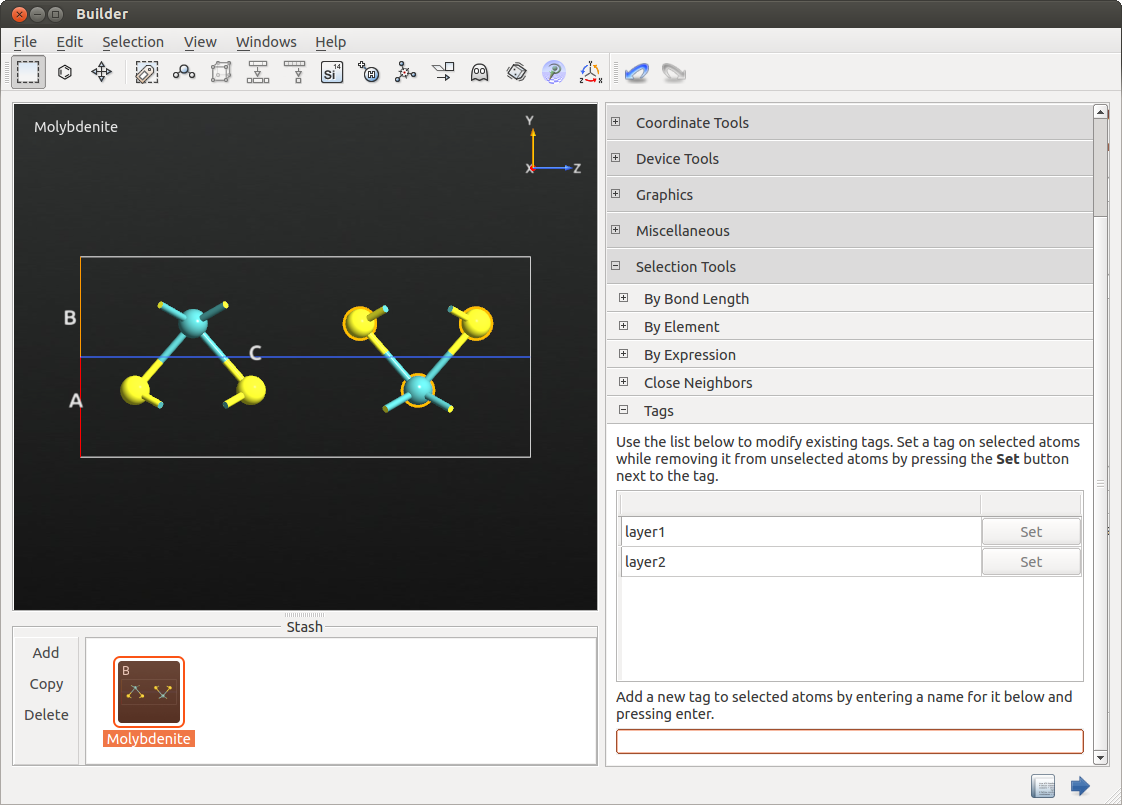
将构型发送到 ScriptGenerator。
在 ScriptGenerator 中添加一个 NewCalculator 和 Optimize ![]() OptimizeGeometry 模块。打开 NewCalculator 设置,选择含有 StillingerWeber_MoS_2013 势的 ATK-ForceField 计算器。取消勾选
OptimizeGeometry 模块。打开 NewCalculator 设置,选择含有 StillingerWeber_MoS_2013 势的 ATK-ForceField 计算器。取消勾选 Save,然后关闭 NewCalculator 控件。打开 OptimizeGeometry 控件,如第一个示例中所述调整几何和应力优化的设置。
将最终脚本发送给到 Editor。
在 python 脚本中,找到定义 Stillinger-Weber 势的行。复制该行,并按以下方式调整这两行:
sw_layer1 = StillingerWeber_MoS_2013(tags='layer1') sw_layer2 = StillingerWeber_MoS_2013(tags='layer2')
这将仅对属于相应标记组的原子间定义每个势。此外,您必须定义只在不同组的硫原子间起作用的 Lennard-Jones 势。这可以通过添加以下代码行来完成:
# Define a new potential for the interlayer interaction. lj_interlayer_potential = TremoloXPotentialSet(name="InterLayerPotential") # Add particle type definitions for both types. lj_interlayer_potential.addParticleType(ParticleType.fromElement(Molybdenum)) lj_interlayer_potential.addParticleType(ParticleType.fromElement(Sulfur, sigma=3.13*Angstrom, epsilon=0.00693*eV)) # Add Lennard-Jones potentials between the sulfur atoms of different layers. lj_interlayer_potential.addPotential(LennardJonesPotential('S', 'S', r_cut=10.0 * Angstrom)) lj_interlayer_potential.setTags(['layer1', 'layer2'])
Lennard-Jones 参数取自参考文献[5]。最后一行确保了 Lennard-Jones 势只在交替层间起作用。
最后,所有三个势的集合必须在 TremoloX 计算器中组合。为完成此操作,请添加以下行:
# Combine all 3 potential sets in a single calculator. calculator = TremoloXCalculator(parameters=[sw_layer1, sw_layer2, lj_interlayer_potential])
完成定义势之后,将计算器添加到块体构型上并执行几何体优化,脚本照常继续。您可以在文件 ↓ Molybite_SW_plus_LJ.py 中找到最终脚本以作比较。
如果您运行脚本,将会获得优化的晶格常数,其与参考文献 [5] 中报道的数据非常相似。
参考
- [1] (1, 2, 3, 4, 5) M Matsui and M. Akaogi: Molecular dynamics simulations of the structural and physical properties of the four polymorphs of TiO2. Mol. Sim. 6, 239 (1991)
- [2] (1, 2) C.J. Fennell and J.D. Gezelter: Is the Ewald summation still necessary? Pairwise alternatives to the accepted standard for long-range electrostatics. J. Chem. Phys. 124, 234104 (2006)
- [3] (1, 2, 3, 4, 5) J.P. Trinastic, R. Hamdan, Y. Wu, L. Zhang, H.-P. Cheng: Unified interatomic potential and enery barrier distributions for amorphous oxides. J. Chem. Phys. 139, 154506 (2013)
- [4] (1, 2, 3) O. N. Kalugin, V. V. Chaban, O. V. Prezhdo: Microscopic Structure and Dynamics of Molecular Liquids and Electrolyte Solutions Confined by Carbon NanoTubes: Molecular Dynamics Simulations. Carbon Nanotubes - Synthesis, Characterization, Applications (2011), Siva Yellampalli (Ed.).
- [5] (1, 2) J.-W. Jiang, H.S. Park, T. Rabczuk: Molecular dynamics simulations of single-layer molybdenum disulphide (MoS2): Stillinger-Weber parameterization, mechanical properties and thermal conductivity J. Appl. Phys. 114, 064307 (2013)