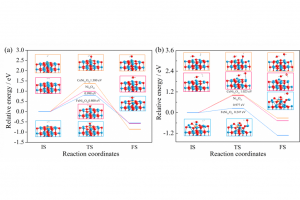
CLC作为一种低能耗、低污染的新型能源利用方法,是实现清洁生产和高效转化的研究热点。CLC过程的关键任务之一是载氧体的反应活性和机理研究。众所周知,燃烧等热化学转化过程的化学反应行为与反应物的结构密切相关。NiO、Fe2O3、钙钛矿和尖晶石材料在CLC中表现出不同的反应行为和机理,这些差异主要来自于各种载氧体的化学组成、颗粒尺寸、表面形态和化学键特征。尽管已经在实验中研究了CLC中各种反应行为,从多尺度分子模拟、反应动力学和材料表征的角度,尚未全面了解Fe/Co掺杂的镍基载氧体的反应行为和机理,仍然面临许多局限性和挑战。 因此,本工作通过多尺度分子模拟和实验结合研究CLC过程中Fe/Co掺杂的镍基载氧体的反应机理和行为。主要工作由三部分组,分别是本征反应机理和性能、非本征反应性能评价、材料表征揭示反应行为。首先,采用多尺度分子模拟研究Fe和Co掺杂的NiO载氧体和H2分子之间的本征反应的反应能垒和机理。使用IGM和EDA研究H2分子与载氧体表面之间相互作用的区域、类型和强度。然后,使用H2-TPR和TGA测试系列Fe和Co掺杂的镍基载氧体的性能,得到转化率和表观活化能。最后,从颗粒尺寸、形貌、表面等材料表征学揭示本征反应和非本征反应的差异。 研究要点一: ReaxFF MD模拟提供了直接证明H2与NiO相互作用生成H2O分子的反应是基元反应的证据。整个CLC过程中截取H2生成H2O的反应机理如图1所示,H2向活性位点扩散,并倾斜吸附在载氧体表面(图1(a))距离约为2 Å。紧接着O原子迅速向H2转移,H-H键断裂的同时形成H…O…H键(图1(b))。形成的不稳定的结构,Ni-O键随即断开生成H2O分子(图1(c))。当脱附完成时,H2O分子的H-O键长分别为1.464和0.960 Å,H…O…H键角约为125°。H2O分子进一步弛豫得到合理的结构,并远离载氧体表面。下面接着使用DFT计算探索反应物、产物和过渡态的结构和能量更准确地描述反应过程。 图 1 截取ReaxFF MD模拟反应过程H2生成H2O的过程(红色球、蓝色球和白色球分别是O、Ni和H原子) 研究要点二: H2吸附在4层2×2的NiO (001)周期性平板模型(如图2(a)所示),镍和氧原子各32个,表示为Ni32O32。考虑Ni原子被Fe/Co掺杂取代的NiO载氧体模型表示为MNi31O32(M=Fe、Co和Ni),如图2(b)所示。H2吸附在MNi31O32载氧体表面的吸附构象分别是H2垂直吸附在M-top、O-top和空位(分别表示为T1、T2和T3),以及H2平行吸附在M—O、M…Ni和O…O桥位(分别表示为P1、P2和P3),其中M=Fe、Co或Ni,如图4-2(c)所示。总共构建18个结构用于研究H2在镍基载氧体表面反应机理。载氧体的连续氧释放反应模型被考虑并表示为Ni32O31、FeNi31O31和CoNi31O31。 图 2计算结构模型:(a) 4层2×2 NiO超胞模型;(b) MNi31O32模型(M=Ni、Fe或Co);(c)吸附位点 研究要点三: 18种模型的Ebinding值范围为-0.236~-0.016 eV。H2在Ni32O32表面的P3构象Ebinding值最负(-0.192 eV),是最佳吸附构型。该结果与ReaxFF MD模拟的结果一致,H2分子倾向于平行吸附在Ni32O32表面的O…O键。P2是最不稳定的构型,Ebinding值为-0.0761 eV。如图3(a)所示,H2吸附在FeNi31O32和CoNi31O32表面的P3构象的Ebinding最负(分别为-0.212和-0.236 eV)是优势吸附构象。该结果与H2吸附Ni32O32表面的优势吸附构象一致。因此,H2分子倾向于平行吸附在Ni32O32、FeNi31O32和CoNi31O32表面的O…O键。除了T1和T3外,随着Fe和Co掺杂,大多数Ebinding值变得更负,这表明Fe/Co掺杂有利于H2在OCs表面上的吸附。 研究要点四: 采用能量分解对H2分子吸附在Ni32O32、FeNi31O32和CoNi31O32表面的稳定吸附构象进行分析。最稳定的吸附模型(P3)的EPauli分别为0.264 eV、0.227和0.251 eV,这表明Fe和Co掺杂减少排斥相互作用表现出显著地稳定效应,这可以有效地活化载氧体的晶格氧。相比之下,Eelstat、Eorb和Edisp在P3吸附构象对总吸引力的贡献比例不同。H2吸附在Ni32O32、FeNi31O32和CoNi31O32表面的P3,Eelstat(38.1、36.1和38.0%)和Edisp(34.3、36.7和35.3%)的贡献几乎相等,并且两种相互作用的贡献都大于Eorb(27.6、27.2和26.7%)。因此,静电和色散相互作用为主,其次是轨道相互作用。这与表现出较弱相互作用的体系不同。Ni32O32、FeNi31O32和CoNi31O32在T2的Edisp的贡献分别增加到65.9、64.9和67.8%,因此色散相互作用成为三个载氧体表面吸引力的主要贡献,Eelstat和Eorb的贡献均低于20%。T1吸附构型中,Ni32O32以Eorb(50.3%)为主,而CoNi31O32以Edisp(53.2%)为主。Eelstat(29.5%)、Eorb(35.9%)和Edisp(34.6%)构成了T3的总吸引力。根据EDA结果,通过不同吸附模型调节静电、轨道和色散相互作用可以有效地增强H2吸附载氧体表面。 研究要点五: 图3(b)总结了CLC中H2与Ni32O32、FeNi31O32和CoNi31O32载氧体的不同反应势能面、反应坐标和反应能垒(ΔEbarrier)。H2在Ni32O32表面的ΔEbarrier值在0.998~1.974 eV。H2在Ni32O32表面的反应,P3的ΔEbarrier最低(0.998 eV),P2最高(1.974 eV);FeNi31O32表面反应的ΔEbarrier值从0.800 eV (P3)到1.614 eV (T2);CoNi31O32表面反应的ΔEbarrier值1.380~2.677 eV。因此,Fe掺杂降低了反应能垒,而Co掺杂相反。FeNi31O32和Ni32O32最佳反应路径都是P3,没有改变最佳反应路径。Co掺杂改变了反应路径,这可能导致CoNi31O32载氧体ΔEbarrier增加的因素之一。除了反应路径和ΔEbarrier值之外,另一个原因可能是Co掺杂导致更负的Ebinding值(较低的IS结合能),使能垒增大。 图3 不同构象的(a) Ebinding和(b) ΔEbarrier○在(a)中表示最负的结合能;(b)中表示能垒最高。△在(a)中表示最大的结合能;在(b)中表示能垒最低。 图4(a)所示H2与Ni32O32、FeNi31O32和CoNi31O32载氧体反应的最低ΔEbarrier值分别为0.998、0.800和1.380 eV。载氧体的本征反应的活性顺序为FeNi31O32>Ni32O32>CoNi31O32。这可能归因于纯Ni32O32中引入掺杂剂导致M—O键和M…Ni(M=Fe或Co)相比完美Ni32O32的Ni—O键和O…O和Ni…Ni键扭曲。H2平行吸附于纯Ni32O32表面的O…O (P3),距离为2.724 Å。H2在FeNi31O32表面,虽然P3是优势吸附构象,但H2略微偏向掺杂的Fe原子。Co掺杂的优势构象H2吸附在掺杂的Co原子上(T1),并且更靠近表面(3.048 Å)。在TS结构中,氧原子分别在Ni、Fe和Co原子上,H2O分子位置保持不变,直到反应完成(FS结构)。Fe和Co掺杂对载氧体中的M—O键(M=Fe、Co或Ni)和晶格氧的影响不同,从而显着改变了反应能垒。图4(b)是Ni32O31、FeNi31O31和CoNi31O31连续释氧过程的反应坐标和路径。Ni32O31、FeNi31O31和CoNi31O31的ΔEbarrier值分别为0.977、0.247和1.023 eV,略低于第一个释氧的值,说明第二个释氧反应更容易。由于Ni32O31、FeNi31O31和CoNi31O31含有氧空位有利于CLC反应,这和之前的报道的一致。 图4最佳势能面的ΔEbarrier和结构 (a) Ni32O32、FeNi31O32和CoNi31O32表面;(b) Ni32O31、FeNi31O31和CoNi31O31表面 结论: […]