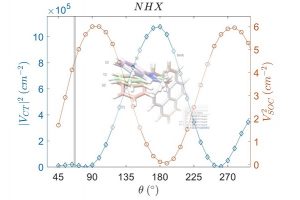
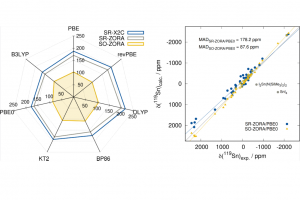
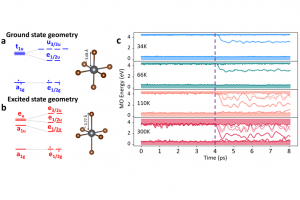
摘要 理论计算得到的电场梯度(EFG),有助于从电子和分子结构的角度解释实验确定的核四极相互作用(NQI)。因此,精确计算EFG是研究许多类型分子结构的核心。在最近的一项工作中,哥本哈根大学的研究人员研究了 CueR 蛋白的模型体系。 针对 CCSD(T) 计算 Cd(SCH3)2 中 Cd(II) 位置的 EFG 的结果,作者测试各种 DFT 泛函后,选择了 BHandHLYP 泛函。使用 AMS 软件中 ADF 模块,评估了使用标量相对论和自旋轨道相对论、基组大小和 CueR 蛋白质模型系统大小对计算结果的影响。从中心 Cd(II) 以外第一个 C–C 键处截断,这样产生的模型体系不足以获得可靠的 EFG 性质,必须采用更大的模型系统来实现可靠的 EFG 计算。非相对论和标量相对论性计算的最大差值为 0.28 a.u.,即 Vzz 的 9%。包含自旋-轨道耦合后,Vzz 进一步增加了 0.05 a.u。从结果来看,很显然自旋-轨道耦合的贡献微不足道,但标量分量至关重要。 使用 BHandHLYP 函数和局部稠密基组(QZ4P、TZ2P 和 DZP 的组合)的自旋轨道耦合计算提供了可靠的结果,并能够对实验数据进行结构解释。 参考文献 Catriona A. O’Shea, Rasmus Fromsejer, Stephan P. A. Sauer, […]