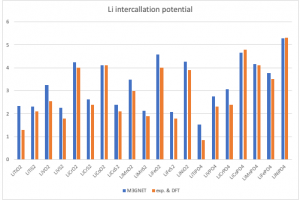
本文实现功能简介 因为 M3GNET 是针对这类锂电池材料结构的能量而训练得到的力场,因此能够很好地、符合预期地再现这些材料中的 Li 嵌入势。下图是 M3GNET 与 DFT 或实验结果的对比: 因此 M3GNET 可以用于筛选未知材料,或快速评估给定成分的 Li 嵌入势。不过 M3GNET 无法很好地描述脱锂前后体积的变化,因为它是基于 PBE(+U) 训练的,并且一些相关材料的结构没有用该理论水平很好地描述(理想情况下,结构应使用 BAND 和SCAN泛函进行计算)。 本文提供了一个基于 PLAMS 的 Python 脚本,可以调用 M3GNET 力场或者基于DFT的 BAND 模块进行计算,对不同材料的Li嵌入势进行筛选。其中 Li 嵌入势的定义,例如对 LinO6Ti3: VLi = [E(LinO6Ti3) -E(O6Ti3)-E(Li)*n]/n 脚本还计算了晶胞体积变化:V(O6Ti3) – V(LinO6Ti3) 脚本的使用 下载压缩文件并解压,得到一个 cif 文件夹以及一个 Python 脚本名为 Li_potential.py。其中 cif 文件夹存放的是样本结构,可以存放多个。筛选时,用户可以基于这些样本结构,分别替换其中的过渡金属和氧元素,然后计算上述两种数据。 用户可以通过如下2行修改过渡元素和替换氧元素的元素列表: tm_list = [‘Ti’,’V’,’Cr’,’Co’,’Mn’,’Fe’,’Ni’] anion_list = [‘O’,’S’] […]