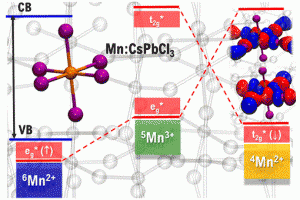
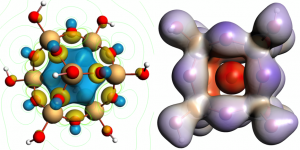
Mn掺杂的铅卤化铅钙钛矿的掺杂发光寿命长,主体激子量子产率高。沙特阿拉伯国王大学Edoardo Mosconi课题组与意大利技术研究院Filippo De Angelis课题组等,通过对APbX3钙钛矿(X=Cl,Br,I)的DFT计算,研究了Mn掺杂钙钛矿敏化掺杂发光过程中,能量和电荷转移的争议问题。 作者定量地模拟了Mn掺杂钙钛矿在不同电荷和自旋状态下的电子结构,将Mn敏化作为钙钛矿组分的函数,对结构/机理进行了分析。该分析的结果,同时支持能量转移机制和电荷转移机制。后者可能更适合于Mn:CsPbCl3,因为它具有较小的能量势垒,并规避了自旋和轨道方面的限制。在电荷转移的情况下,决定掺杂发光量子产率的一个重要因素是中间氧化物种的能量,而带隙共振可以很好地解释能量转移。这两个方面由钙钛矿主体的带边能量控制,而这又可以被卤化物X所调制。 参考文献: Damiano Ricciarelli, Daniele Meggiolaro, Paola Belanzoni, Asma A. Alothman, Edoardo Mosconi*, and Filippo De Angelis*, Energy vs Charge Transfer in Manganese-Doped Lead Halide Perovskites, ACS Energy Lett. 2021, 6, XXX, 1869–1878