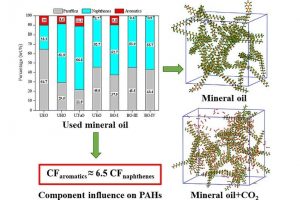
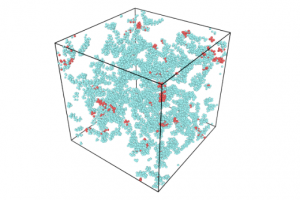
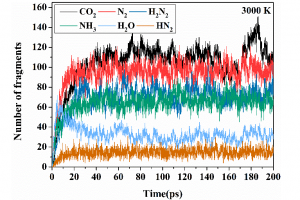
摘要 通过热解回收废旧矿物油是一种灵活有效的方法。然而,在这个过程中可能会产生具有高毒性的多环芳烃(PAHs)。本工作试图通过ReaxFF分子动力学(MD)模拟揭示矿物油热解过程中矿物油的演化过程和多环芳烃的形成机制。此外还探讨了加热速率(10 K/ps、100 K/ps和~1000 K/ps)、温度(2200~3200 K)、矿物油成分(环烷烃和芳烃含量)和大气(CO2)影响多环芳烃形成的原理。观察到矿物油的两阶段热解演化:第一阶段分解,第二阶段聚合反应。高温可以使热分解快速转化为聚合反应阶段。通过跟踪关键中间体/产物和芳香结构的演变发现,在矿物油热解过程中,氢提取-乙烯基自由基加成(HAVA)反应主导了PAHs的形成。此外,连接在碳簇上的支链基团的缩聚环化对大的PAHs的形成做出了相当大的贡献。乙烯基加成和缩聚脱氢是矿物油热解过程中PAHs形成的两个标志性反应。矿物油中芳烃组分对多环芳烃形成的贡献约为环烷组分的6.5倍。CO2可以通过中间体/产物的氧化以及随后的乙烯基/乙炔加成反应对脱氢的抑制来减少PAHs的形成。 参考文献 Formation mechanism of polycyclic aromatic hydrocarbons during mineral oil pyrolysis: A ReaxFF molecular dynamics study, Linlin Xu, Gan Wan, Lushi Sun, Li Lin, Fuel, 2024, 131175, DOI: 10.1016/j.fuel.2024.131175