
1. 计算模型和方法

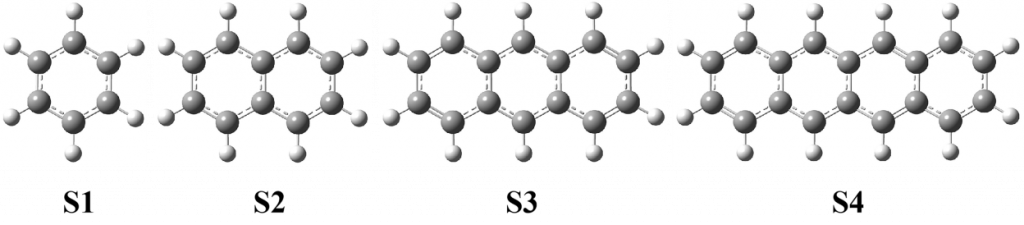
文中选取化学链技术中最常见的Fe2O3载氧体。构建α-Fe2O3(001)低指数晶面。以Fe-O3-Fe…为反应表面的稳定结构扩胞得到初步铁基载氧体颗粒模型。然后将多环芳烃放到Fe2O3载氧体表面,然后再进行5×5超胞。ReaxFF MD模拟的周期性盒子设定为100 Å×100 Å×50 Å。如图所示,Fe2O3载氧体总共8层16000个原子,其中氧原子9600个,Fe原子6400个。载氧体上面共有25个多环芳烃。多环芳烃选择了固体燃料中具有代表性的4种多环芳烃,这些多环芳烃在许多煤分子结构是常见的。
主要对CLC过程反应器内Fe2O3和多环芳烃体系反应过程ReaxFF MD模拟。计算采用郑等开发的包含C/H/O/Fe等元素的ReaxFF力场参数,以及NVT正则系综分子动力学(NVT-MD)方法进行计算。计算中,步长为0.25 fs,温度由阻尼常数为0.1 ps的Berendsen恒温器控制。模拟反应温度分别为2000、2500、3000、3500、4000 K,探究不同温度Fe2O3载氧体和多环芳烃反应特性。反应前计算模型在450 K进行了40000步非反应(Non-reaction)弛豫过程,使其处于平衡状态。ReaxFF MD模拟过程累积1000000步,总时长250 ps。
2. 结果讨论
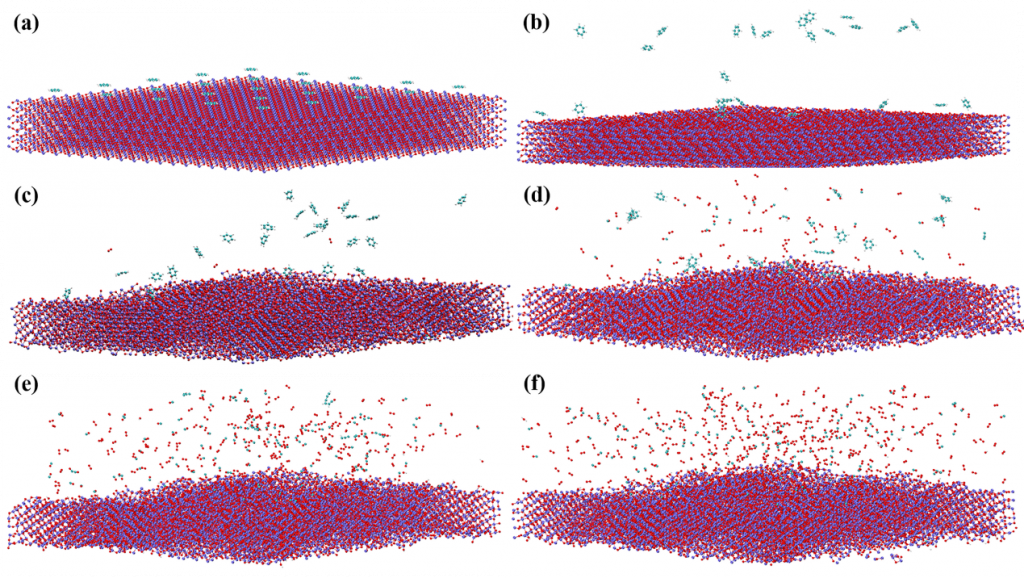
图3是多环芳烃在Fe2O3载氧体表面反应的可视化过程。根据截取的0、25 、 75 、125、175、250ps时CLC体系实时图片,燃烧过程可分为四个阶段。通过上述化学链燃烧不同阶段的反应物和产物分析发现,这些多环芳烃分子和铁基载氧体化学链燃烧过程可分为四个阶段。第一个阶段属于反应前阶段,主要发生载氧体颗粒和燃料分子的结构弛豫和扩散。该过程使得体系中载氧体颗粒和燃料分子中化学键呈现键合-解离-键合的动态平衡特征。这将有助于它们在后续过程中参与反应。第二阶段属于初始反应阶段。此阶段多环芳烃自身开始逐步发生热裂解反应,生成自由基和含碳碎片。同时,部分燃料分子或反应碎片与载氧体表面的晶格氧距离靠近,形成有效相互作用,进而形成化学键连。载氧体开始发生反应逐步释氧 。第三阶段属于剧烈燃烧反应阶段。此时多环芳烃在高温环境中大量裂解成分子碎片。载氧体大量释氧同步发生。整个化学链体系呈现剧烈燃烧反应的特征。第四阶段属于最终燃烧阶段。此时大部分多环芳烃已裂解成碎片。主要发生的反应为大量含碳碎片中间体物种的氧化燃烧。该过程生成大量典型燃烧最终产物,如CO,CO2,H2O等。
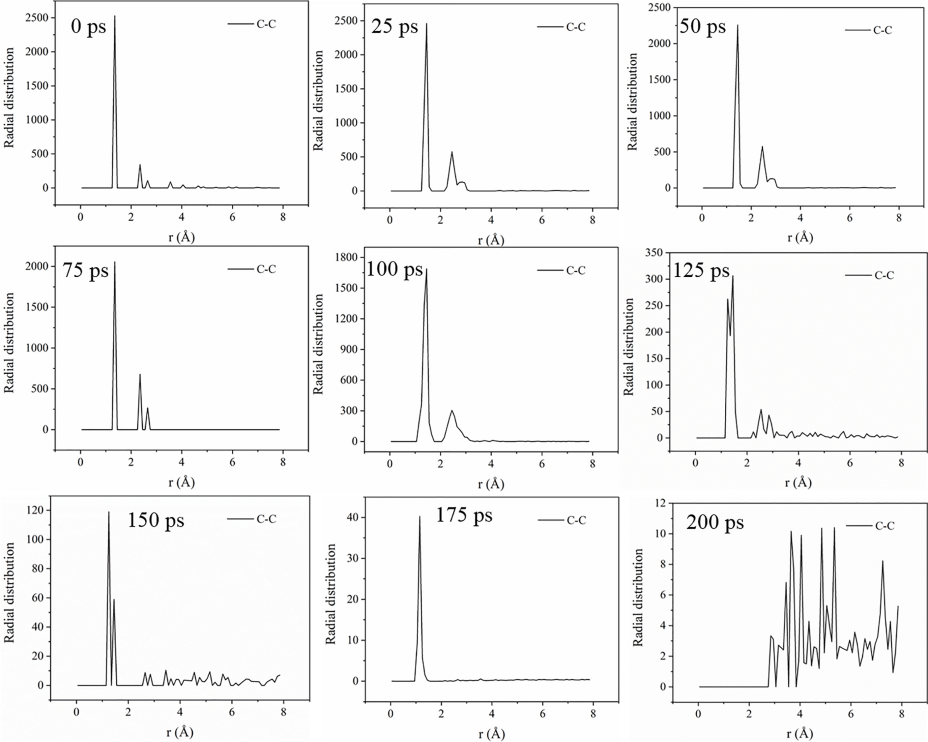
为了系统了解不同芳烃燃料分子在化学链燃烧过程中分子结构变化,对燃料分子S1在CLC过程中不同反应时间体系的C…C相互作用距离进行了径向分布函数(RDF)分析。RDF在约1.4 Å 处的峰可反映体系中C-C键的数量变化,并表征芳烃燃料分子的热分解反应动力学。
在50 ps时1.4 Å处峰的强度仍2500。这表明此时燃料分子中C-C数量不变,燃料分子尚未解离。在75/ 100/ 125/ 150/ 175 ps时,RDF中1.4 Å处峰的强度分别为2000,1700,300,120和40。这表明反应过程中燃料分子中C-C数量减少,燃料分子逐步解离发生燃烧反应。通过RDF和 C-C键数量变化分析,可以很好地反映上述化学链燃烧反应的四个阶段。

研究发现,CLC反应温度对燃料分子参与化学链燃烧反应影响显著。燃料分子随CLC温度升高呈现参与反应提前现象。从燃料分子反应数量可发现,随着CLC温度升高,芳烃分子参与反应越早。例如,S1分子在3500 K时,在50 ps开始反应分解,而2000/2500/3000 K时则分别为62.5,62.5,50 ps。因此,S1分子在3500 K的反应分别比2000/2500/3000 K提前了12.5,12.5,0 ps。同时还发现,CLC温度对于燃料分子分解时间具有影响。分析燃料分子反应数量可发现,随着CLC温度升高,芳烃分子全部参与CLC反应耗时减少。例如,S1分子在3500 K时,在50 ps开始反应分解, 137 ps体系无S1分子。S1分子全部参与反应并分解总耗时87 ps。而相应的,2000/2500/3000 K的CLC过程中S1分子全部参与反应并分解总耗时分别为125、113和100 ps。因此,S1分子在3500 K的反应比2000/2500/3000 K分别提前了38,26,13 ps。燃料分子S2-S4在CLC过程中不同温度下反应数量变化也可获得相同结论。可见,高温利于燃料分子在CLC过程中的转化。这与燃烧反应中升高温度可以加快反应速率的反应动力学常识一致。

这里以S1燃料分子的反应为例进行分析。在3500 K下CO在约75 ps开始产生。100-175 ps期间CO数量迅速增加,CO从5个增加到130个,直至200 ps后CO趋于稳定增加到132个。所以CO 的生成规律符合前面的四个反应阶段。研究发现,CO的释放数量并非CLC温度越高越有利。CLC模拟温度在3000 K下250 ps时ReaxFF MD产生的 CO分子数量最多(134 个)。在2000,2500,3000 K下250 ps时生成CO的数量为83,113,134个。3000-4000 K下生成CO数量非常接近,分别为134,134,130个。研究发现,模拟反应温度为4000 K时,CO数量在212-250 ps有下降趋势,CO数量从141个下降到130个。可能导致该现象的潜在原因是CO继续氧化向CO2转换所致。
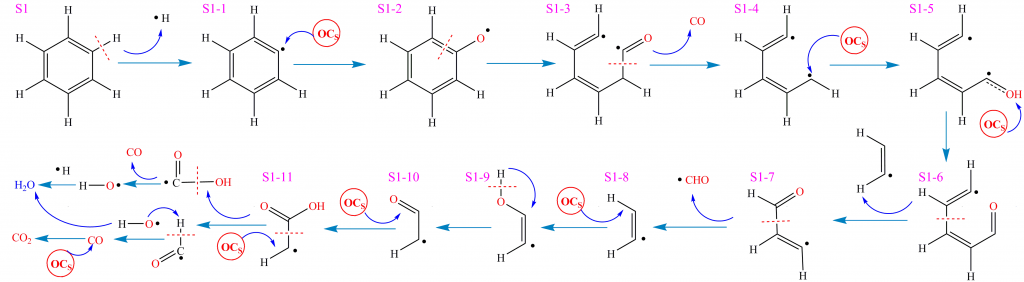
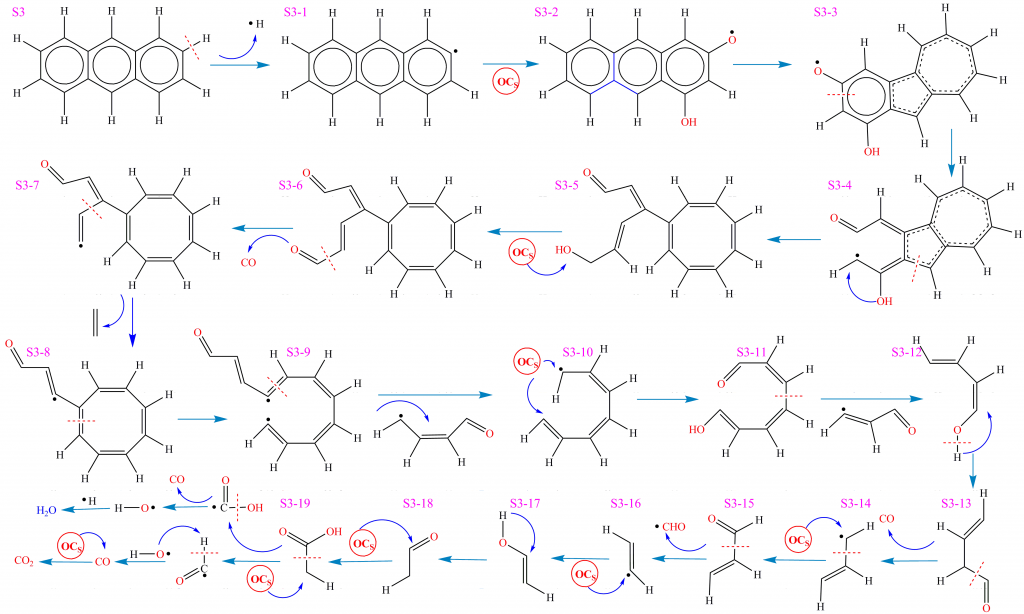
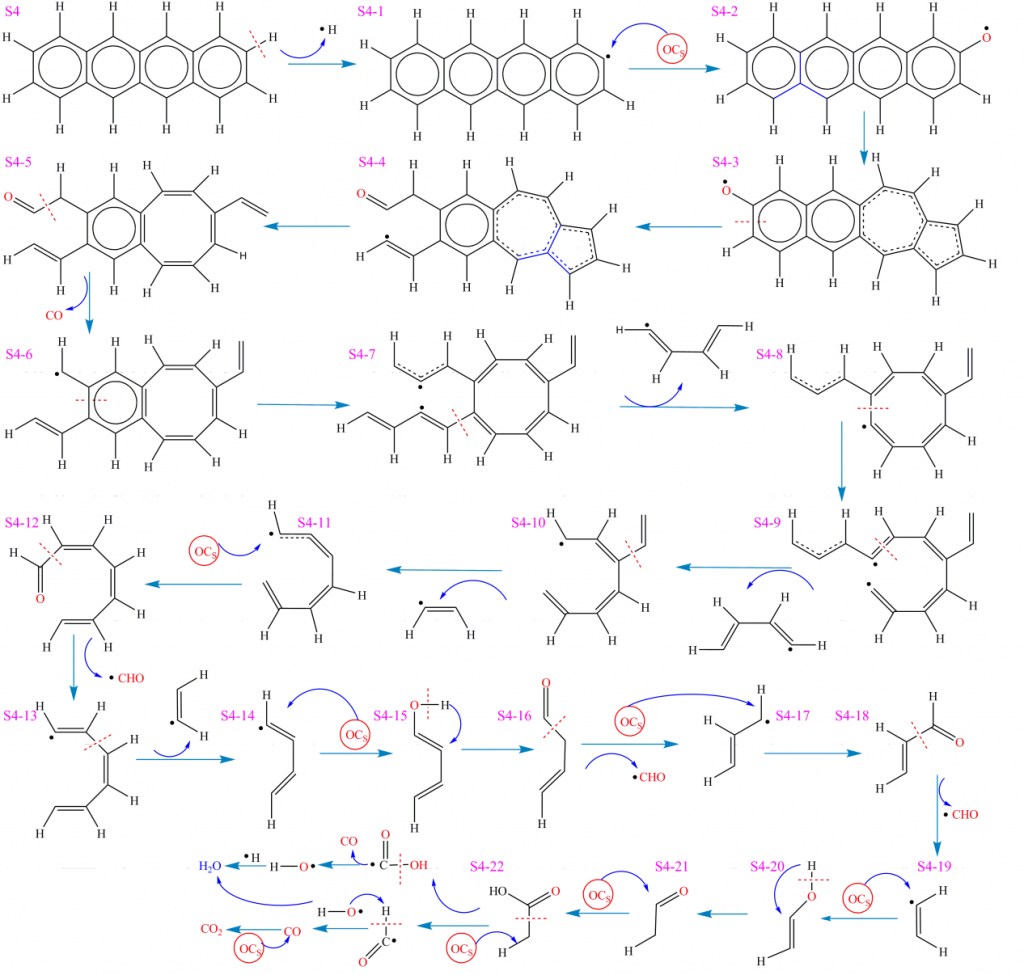
如图是根据ReaxFF MD可视化结果获得的S1-S4分子在CLC过程中复杂环境界面的反应路径和网络。首先对S1分子的反应进行分析。S1分子初始反应阶段涉及热裂解与氧化反应的竞争,存在多种不同的热化学反应方式。在主反应路径中,S1分子中C-H断裂(35-38)生成C6H5•(物种S1-1)和H•自由基。C6H5•与载氧体中晶格氧后解离,被氧化为C6H5O•(物种S1-2) 。C6H5O•经开环反应生成(Z)-hexa-3,5-dienal(物种S1-3),进一步解离生成CO和(Z)-penta-1,3-diene(物种S1-4)。S1-4继续结合晶格氧经过多步反应并发生片段解离,分别释放ethene(物种S1-8)和acrylaldehyde(物种S1-7)。S1-7解离后释放•CHO自由基和ethene(物种S1-8)。此后,ethene继续被晶格氧进攻生成ethenol(物种S1-9)。该结构不稳定,经重排反应转化为acetaldehyde(物种S1-10)。acetaldehyde被载氧体氧化为acetic acid(物种S1-11)并进一步解离生成•CHO和•COOH。•CHO和•OH自由基反应生成H2O和CO,CO被载氧体继续氧化为CO2。另一方面,•COOH解离生成CO和•OH自由基,后者和H•结合生成H2O。

本文反应力场分子动力学计算,采用 AMS(Amsterdam Modeling Suite )中 ReaxFF模块 完成。
3. 文章信息
Exploring depolymerization mechanism and complex reaction networks of aromatic structures in chemical looping combustion via ReaxFF MD simulations
Liangliang Meng1#, Ying Zhu2#, Meilin Zhu3, Ge Wu1, Wenqian Guo1, Chang Geng1, Na Li1, Rou Feng1, Hui Zhang1, Qingjie Guo1, and Hongcun Bai1*
1 State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering, College of Chemistry and Chemical Engineering, Ningxia University, Yinchuan 750021, Ningxia, China
2 School of Pharmaceutical Sciences, Hunan University of Medicine, Huaihua 418000, Hunan, China
3 College of Basic Medical Sciences, Ningxia Medical University, Yinchuan 750004, China
# These authors contributed equally to this work
* Corresponding author: Dr. H. Bai (E-mail: hongcunbai@gmail.com, hongcunbai@nxu.edu.cn; Tel.: +86- 951- 2062328; Fax: +86-951-2062323)
期刊:Journal of the Energy Institute (IF 6.47)