
这是本文档旧的修订版!
NiSi2-Si界面
在这篇教程中,我们研究一个掺杂的 <chem>NiSi2</chem>-Si 表面接触的输运性质。我们将会学习到:
- 构建一个掺杂的表面接触结构,计算它的输运性质;
- 学习如何适当掺杂一个真实的半导体器件;
- 利用 VNL,分析结构的电学性质、电子密度和态密度。
构建一个NiSi2-Si器件
- 生成一个空的项目,并打开
 Builder.
Builder. - 首先要做的是,生成一个用LDA优化过的<chem>NiSi2</chem>-Si的晶体结构生成一个器件。优化过的晶体结构文件可在此处下载:
- Interface.py
# ------------------------------------------------------------- # Bulk Configuration # ------------------------------------------------------------- # Set up lattice vector_a = [3.87406675195, 0.0, 0.0]*Angstrom vector_b = [0.0, 3.87406675195, 0.0]*Angstrom vector_c = [0.0, 0.0, 32.9399103335]*Angstrom lattice = UnitCell(vector_a, vector_b, vector_c) # Define elements elements = [Nickel, Silicon, Silicon, Nickel, Silicon, Silicon, Nickel, Silicon, Silicon, Nickel, Silicon, Silicon, Nickel, Silicon, Silicon, Nickel, Silicon, Silicon, Silicon, Silicon, Silicon, Silicon, Silicon, Silicon, Silicon, Silicon, Silicon, Silicon, Silicon, Silicon] # Define coordinates fractional_coordinates = [[ 0. , 0.5 , 0.041751877231], [ 0. , 0. , 0.083503754463], [ 0.5 , 0.5 , 0.083503754463], [ 0.5 , 0. , 0.125255631694], [ 0. , 0. , 0.167007508926], [ 0.5 , 0.5 , 0.167007508926], [ 0. , 0.5 , 0.208759386157], [ 0. , 0. , 0.250511263389], [ 0.5 , 0.5 , 0.250511263389], [ 0.5 , 0. , 0.29226314062 ], [ 0. , 0. , 0.334015017852], [ 0.5 , 0.5 , 0.334015017852], [ 0. , 0.5 , 0.375766895083], [ 0. , 0. , 0.417518772314], [ 0.5 , 0.5 , 0.417518772314], [ 0.5 , 0. , 0.459270649546], [ 0. , 0. , 0.501022526777], [ 0.5 , 0.5 , 0.501022526777], [ 0. , 0.5 , 0.542603982879], [ 0.5 , 0.5 , 0.584185438981], [ 0.5 , 0. , 0.625766895083], [ 0. , 0. , 0.667348351185], [ 0. , 0.5 , 0.708929807287], [ 0.5 , 0.5 , 0.750511263389], [ 0.5 , 0. , 0.79209271949 ], [ 0. , 0. , 0.833674175593], [ 0. , 0.5 , 0.875255631694], [ 0.5 , 0.5 , 0.916837087796], [ 0.5 , 0. , 0.958418543898], [ 0. , 0. , 1. ]] # Set up configuration bulk_configuration = BulkConfiguration( bravais_lattice=lattice, elements=elements, fractional_coordinates=fractional_coordinates ) # Add tags bulk_configuration.addTags('Left Interface', [ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17]) bulk_configuration.addTags('Right Interface', [18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29])
注意 可以从以下几篇教程中学习如何建立表面接触结构:
- 在Stash面板处,用Add ‣ From Files将下载下来的结构导入到Stash面板。
- 利用右侧的Device Tools ‣ Device From Bulk,生成一个器件。
- 不需要修改电极的长度,但是需要把左右电极长度加大到100%。

屏蔽区域
如何确定掺杂浓度?原则上我们可以用实验值。然而,我们必须对另一个量十分谨慎:耗尽层(译注:又称势垒区)宽度,经常用$W_D$ 表示,和半导体的屏蔽长度有关。 耗尽层宽度和以下几个因素有关:
- 半导体的介电常数
- 掺杂浓度(载流子数目)
- 界面处的势垒
- 偏压
针对金属半导体结,$W_D$ 的取值与掺杂浓度 $N$ 的关系由下图给出(参考文献:[BKasper 2005] pag. 119)

为了使模拟达到合理的目标,我们需要确保硅在中心区域的长度大于耗尽层的宽度。在这篇教程里,我们掺杂的浓度范围为$10^{19} cm^{-3}$到$10^{20} cm^{-3}$。这意味着硅的长度要达到30Å到100Å。我们可以通过将 Electrostatic Difference Potential 作图检查长度是否足够(后面会详细解释)。
注意 金属内的屏蔽长度远小于半导体,一般只需要几个原子层。因此,NiSi2的厚度可以远小于硅层的厚度。
注意
在本教程中,我们将会使用一个较高浓度的掺杂。器件的输运性质与低浓度掺杂(如$10^{17} cm^{-3}$、$10^{18} cm^{-3}$)的性质完全不同。模拟一个低浓度掺杂的器件需要构建更长的器件。
延长中心区域长度
- 在Stash面板处选中器件结构
- 在右侧面板中,点开Device Tools ‣ Central Region size,点击Right一行的加号,增大电极长度至100%。

- 发送结构至Script Generator窗口。
建立无掺杂器件的计算
把结构发送至 Script Generator 窗口后,我们接下来就需要设置计算参数了。
 New Calculator,双击添加并修改以下参数:
New Calculator,双击添加并修改以下参数:- 交换关联势选择MGGA
- K点选择 $5\times5\times100$
- 基组选择“SingleZetaPolarized”
- 对于 MGGA 中的参数 c,在 Editor 窗口中做如下需改:
exchange_correlation = MGGA.TB09LDA(c=1.09368541059162)
注意 对c参数,我们是通过单点计算提前获取的。如果想了解更多相关内容以及如何获取c参数,可以参考这篇教程《MetaGGA和二维受限的砷化铟》。同时,我们也比较 LDA 和 MGGA 的能带图,如下图所示:
 图1:利用LDA(左)和MGGA(右)两种方法,所得到的硅晶体结构的能带图
图1:利用LDA(左)和MGGA(右)两种方法,所得到的硅晶体结构的能带图
从图中可以看出,能带结构是非常接近的,但是MGGA的带隙更接近于实验值(1.17eV)。
 Analysis。在弹出的选项中,添加以下内容,并修改相应的参数:
Analysis。在弹出的选项中,添加以下内容,并修改相应的参数:- ElectrostaticDifferencePotentials
- ElectronDensity
- ElectronDifferenceDensity
- ProjectedLocalDensityOfStates
- K点设置为 $25\times25$
- 能量点设置为201
- Method选择”Device DOS”

- DeviceDensityOfStates
- K点设置为 $25\times25$
- 能量点设置为201

3.修改输出文件名字为:device_undoped.nc,并保存脚本为 device_undoped.py. Script Generator 窗口显示如下:

4.发送脚本至Job Manager,并运行。这个计算调用 4 个进程,大概需要花费 2 个小时的时间。其中进程计算 DOS 大约需要 4 个小时。
注意
计算 DOS 时需要花费大量的时间,尤其是采用较大的k点时。我们也可以先选取一个较小的 k 点,如 $15\times15$。我们也可以利用 Analysis from files,稍后计算 DOS。
掺杂器件
1.在 Stash 面板,复制并粘贴器件结构,重命名为 ”n_doped_1e19”
2.用鼠标绘制出一个矩形,选中半导体原子
3.点开 Miscellaneous ‣ Doping

4.在弹出的 Doping 面板中,如图设置相应参数。

5.关闭 Doping 面板,把结构发送至 Script Generator 窗口。
6.设置相应的计算参数。
7.重复相同的步骤,掺杂一个$p$型半导体(浓度为 $10^{19}cm^{-3}$ 和 $10^{20}cm^{-3}$ )
分析结果
计算完成,我们就可以分析计算出来的几组数据了。
静电电位差 Electrostatic difference potential
静电势差(EDP)表征了价电子密度的静电势与原子价电子密度算术叠加的静电势的差异。(详情参看手册: ElectrostaticDifferencePotential)。通过分析沿c方向EDP的平均值,可以判定器件是否足够长——通过判断电位降是否分布在整个电极区域内。
- 在 LabFloor 界面,按住 Ctrl,选中四个 Electrostatic Difference Potentials 项目,点击右侧 1D Projector 工具。

- 在打开的窗口中,如图绘制三条沿 C 方向的平均势能。在图中,点击右键修改图例


从图中可以看出,未掺杂和掺杂浓度为 $10^{19}cm^{-3}$ 的器件长度明显不够(后面会详细计算掺杂浓度为 $10^{19}cm^{-3}$ 的更长器件)。通过分析 EDP,掺杂浓度为 $10^{20}cm^{-3}$ 的$p$型掺杂是唯一在电极扩展层处(靠近 c=1 的部分)显示出单调性的结果。界面位置大概位于 c=0.35 附近,尽管这已经足够说明硅原子和镍原子在这个坐标处结合,但是这个界面并不是一个无限薄的 2D 结构,而是一个有厚度的器件。
电子密度差 Electron Difference Density
电子密度差(EDD)是自洽层电子密度和原子价电子层电子密度叠加的差异。 1.利用脚本(edp_macro_avg.py)绘制 EDD 的平均值。 2.在终端中输入以下命令:
atkpython edp_macro_avg.py file.nc
此处file.nc是包含器件计算的输出文件。
图2:未参杂器件(左)和n型掺杂器件(右)的EDD的宏观平均值

这两幅图表明界面偶极子的存在,尽管这个改变在掺杂半导体中非常小。
电子差异密度(掺杂-未掺杂)
我们可以通过以下几个方面分析两种结构的电子密度的不同:
3D分析
- 在LabFloor处,选择未掺杂器件和n型掺杂( $10^{19}cm^{-3}$ )器件的 ElectronDensity
- 点击LabFloor左侧的Grid Operation
- 在公式编辑器中,输入B1-A1,点击View绘制电子差异密度。
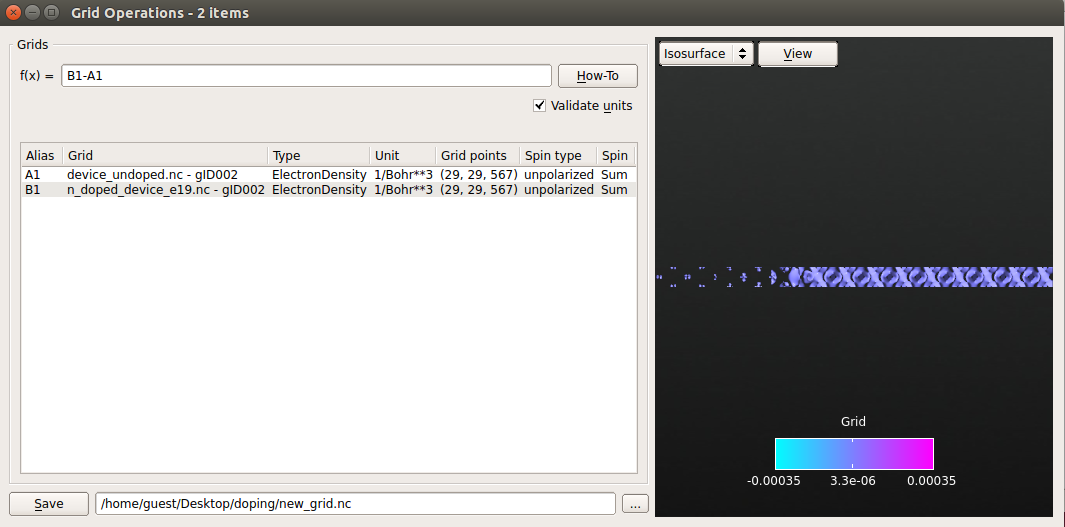
- 将结果保存在 new_grid.nc文件中

1D分析
利用提供的脚本绘制n型半导体和未掺杂半导体的宏观平均差异。
- 打开脚本,将下面这一行:
ax.set_ylabel(r'EDD ($\AA^{-1}$)', fontsize=20)修改为:
ax.set_ylabel(r'B1-A1 ($\AA^{-1}$)', fontsize=20) - 输入以下内容运行脚本:
atkpython edp_macro_avg.py new_grid.nc

这幅图绘制了n型掺杂(B1)和为掺杂(A1)器件,电子密度的不同。在半导体区域,B1-A1>0$e^{-}$$Å^{-}$,这是由于在n型半导体中,硅原子被掺杂了额外的电子。此外,在接触处,B1-A1的值增长,可以理解为由于掺杂界面偶极子发生了改变。
态密度(DOS)分析
在这篇教程中,我们分析两种DOS。
Projected Local Density Of States
- 从 LabFloor 处选择未掺杂器件的 Projected Local Density Of States
- 点击 Projected Local Density Of States
 将会得到下图:
将会得到下图:
 横坐标为沿 c 方向中心区域的笛卡尔坐标。界面位于 27 Å。纵坐标为相对费米能的能量。颜色表征 DOS 的大小。
从图中可以明显看出:
横坐标为沿 c 方向中心区域的笛卡尔坐标。界面位于 27 Å。纵坐标为相对费米能的能量。颜色表征 DOS 的大小。
从图中可以明显看出:
- 器件左侧为金属性,NiSi2;
- 右侧是有带隙的半导体,为未掺杂的 Si;
- 在硅原子区域内,在金属与半导体接触处,有一个较宽的带隙区域,称之为 metal-induced gap states(MIGS)
- 利用相同的方法,计算n型掺杂的local DOS。

从图中可以看出,由于掺杂,费米能级位于带隙的顶部。
Device Density Of States
利用DDOS可以分析制定到原子的态密度。
- 在 LabFoor 选择Device Density Of States。
- 点击 2D Plot,将会弹出2D Plot窗口。
- ( 在3D结构中,选择表面接触的几个原子,将会绘制出DOS图。
 2D图表征了选中原子的DOS。从图中可以看出费米能级附近MIGS的存在。
2D图表征了选中原子的DOS。从图中可以看出费米能级附近MIGS的存在。
- 选择靠近电极处的硅原子:

从图中可以看到,靠近硅电极,MIGS区域消失,并且,电极结构出现了一个带隙,与硅晶体的带隙相似,其结果与Local DOS的结果相吻合。
有限偏压计算
在这一部分,我们将会计算n型掺杂($10^{19}cm^{-3}$)的IV特性曲线。正如我们提到的,掺杂的器件并不是足够长。因此,我们将会用一个100 Å长的器件进行计算。我们利用教程延长中心区域长度增长长度。也可以直接下载结构脚本 long_ndoped_e19_ivcurve.py。
设置IV计算参数
- 发送结构至 Script Generator ,并添加一下内容:
- New Calculator:设置0V参数
- IVcurve:如图设置参数:

这些参数设置的要点:
- 偏压:设置了13个偏压,Vbias= 0.0, -0.05, -0.1, -0.15, -0.2, -0.25, -0.3, 0.05, 0.1, 0.15, 0.2, 0.25 和 0.3 V,Vbias定义为 VL - VR。
注意 每个计算都利用前一次结构自洽的结果,这将极大的提高收敛速度。
- 能量:这个部分指定了每一次偏压下,透射谱需要计算的能量区域和区域内的点数。
警告 能量范围对于计算IV曲线非常重要,详情见http://docs.quantumwise.com/tutorials/nisi2-si.html#nisi2-si-2和http://docs.quantumwise.com/tutorials/nisi2-si.html#nisi2-si-5部分。
- K点:k点取值用来计算透射谱,仅针对透射。一般情况下,计算透射的k点比在自洽计算中所使用的要更些。
警告 K点的取值将会影响到电流!在本教程中,我们选择$13 \times 13$,使透射谱收敛,具体见本教程的相应部分。
- IO file name: 如果要计算多条电流曲线,改变默认的文件名。这个文件将会包含所有的结构信息。正如本教程的相应部分,可以用这个文件进行以下计算:
- 重新计算所有内容,或者计算不同参数的透射谱(k点、能量范围等)
- 计算任意偏压下的其他性质,如DOS、电子密度等
- 拓展偏压范围
- 设定好所有参数后,将脚本发送至 Job Manager,即可运行计算。这个计算在 4 个进程并行的条件下耗时 10 个小时。
透射谱收敛
两个重要参数将会影响透射的收敛:
- 能量点数:这个参数可以用来协调计算精度和所需要的计算资源。理想情况下,透射谱的范围应该至少覆盖偏压范围。本篇教程中,至少应该在0.1eV~0.4eV之间。但是更大的对称区域将会更安全。
- K点:这个参数对透射谱一样有着重要的作用。正如之前提到的,我们需要设置比DFT计算更大的k点,因为计算格林函数比计算电子密度的傅里叶变换更依赖于k点的设置。下图绘制了对于n型掺杂,掺杂浓度为$10^{20}cm^{-3}$,负偏压下,电流随k点取值不同的变化情况。一般来说,需要考虑足够的k点才能使得电流在偏压窗口内收敛。

从图中可以看出,k点选择$13\times13$时,电流就已经非常接近$25\times25$。
警告 这是针对掺杂浓度为$10^{20}cm^{-3}$,n型掺杂的评估,对于浓度为$10^{19}cm^{-3}$的评估,需要另外计算。
电流分析
等到IV曲线计算完了之后,我们可以在LabFloor面板处找看到。
- 选择IVCurve,点击IV-Plot.
- 我们将会在IV-Plot窗口中看到IV曲线。点击Additional Plots,我们将会得到dI/dV、透射谱、谱电流的图像。

重点 谱电流由以下公式给出: $I(E)=e/h[T(E,\mu_R,\mu_L)(f(E-\mu_R)/(k_BT_R)-f(E-\mu_L)/(k_BT_L))]$ 这是综合给定偏压下的数据。这将很好地处理非零偏压下,估算光电流透射谱所需的能量范围。可以参考手册TransmissionSpectrum查看ATK是如何计算电流的。
注意 我们可以在图片任意位置右键导出数据,方便我们后期处理。
有限偏压的分析
正如我们已经提到的,利用自洽结构文件(保存在ivcurve_selfconsistent_configurations.nc 文件中),可以对任意偏压的进行分析。在这部分,我们将会展示如何利用这个文件,并且如何设置脚本对一个结构进行额外的分析。
单自洽结构的分析
1.选择一个自洽结构,点开General info。在面板中,我们可以看到自洽结构的数据。一旦确定需要的结构文件,我们就可以进行设置脚本了

2.转到Script Genera窗口,添加Analysis from File 模块
3.双击 Analysis from File ,在弹出的窗口中插入NC文件,和自洽结构的Object ID。

4.添加所要分析的模块,并设置相应参数。发送脚本至Job Manager。
多个自洽结构分析
这里我们将会展示如何利用自洽结构文件,计算不同偏压下的EDP。
1.利用General info确认需要的自洽结构文件。
2.利用提供的脚本(loop.py),分析多个自洽结构。在脚本里id_list输入需要计算的自洽结构的Object IDs。
id_list = [ 'ivcurve000', 'ivcurve001', 'ivcurve002', 'ivcurve003', 'ivcurve004', 'ivcurve005', ] for id_item in id_list: # Read the DeviceConfiguration configuration = nlread('ivcurve_selfconsistent_configurations.nc', object_id="%s" %id_item)[0] # ------------------------------------------------------------- # Electrostatic Difference Potential # ------------------------------------------------------------- electrostatic_difference_potential = ElectrostaticDifferencePotential(configuration) nlsave('analysis.nc', electrostatic_difference_potential)
3.发送脚本至Job Manager,并运行计算。
分析IV曲线
在这一部分,我们将会分析几个不同偏压下的EDP和-0.3V与0.3V下的LDOS。我们可以模仿上面介绍的步骤进行计算,同时也可以下载脚本:l_ndoped_e19_EDP.py,l_ndoped_e19_DDOS.py。 得到的电流曲线可以分为两个区域:“Forward Bias”($V_{bias}>0V$)以及“Reverse Bias” ($V_{bias}<0V$)。

从图中可以看出,反向偏压电流低于正向偏压电流。这可以利用LDOS进行解释。
下图为在-0.3eV下的LDOS。

在反向偏压条件下,左电极的化学势高于右电极的化学势。因此,此处将会有一个从左电极到右电极的电子净流动,导致电流有一个反向传播。但是由于阻挡层的存在,电子在界面交界处不得不越过一个势垒,导致了电流非常低。
接下来我们计算了0.3V下的LDOS。
 在正向偏压条件下,我们可以看出右电极的化学势要高于左电极。因此,此处将会有一个从右电极到左电极的电子净流动,导致电流有一个反向传播。然而,在不同于反向偏压,此处电子不需要越过任何势垒,因此电流比较高。
在正向偏压条件下,我们可以看出右电极的化学势要高于左电极。因此,此处将会有一个从右电极到左电极的电子净流动,导致电流有一个反向传播。然而,在不同于反向偏压,此处电子不需要越过任何势垒,因此电流比较高。
最后,在下图中,我们计算了不同偏压下,较长n型掺杂器件沿C方向的EDP:
 发现偏压同样也影响了EDP和阻挡层的宽度。
发现偏压同样也影响了EDP和阻挡层的宽度。
警告 在上图中,EDP与$(\mu_L+\mu_R)/2$有关,这是在计算中默认的定义。为了更直观,我们需要绘制一个和$\mu_L$相关的EDP。
参考文献
[bKP05] E. Kasper and D. J. Paul. Silicon Quantum Integrated Circuits. Springer-Verlag Berlin Heidelberg, 2005. doi:10.1007/b137494. (中文翻译:闫强)