
文献重现:锌酞菁的基态与激发态计算(第二部分)
参考文献:Ground and Excited States of Zinc Phthalocyanine Studied by Density Functional Methods, G. Ricciardi and A. Rosa, E. J. Baerends, J. Phys. Chem. A 2001, 105, 5242-5254
激发态计算
参数设置
采用优化好的锌酞菁分子结构(即前文中最后一步计算采用的结构)进行计算:
ADFinput支持比较灵活的分子拷贝、粘贴操作,例如:
将前一次计算的结构,从*.run文件

或者在上一次计算使用的ADFinput界面里面按快捷键CTRL A(全选),此时ADFinput窗口,所有的原子都被选中,呈高亮状态(当然如果我们希望复制的是一部分原子也是可以的),然后CTRL C(复制)
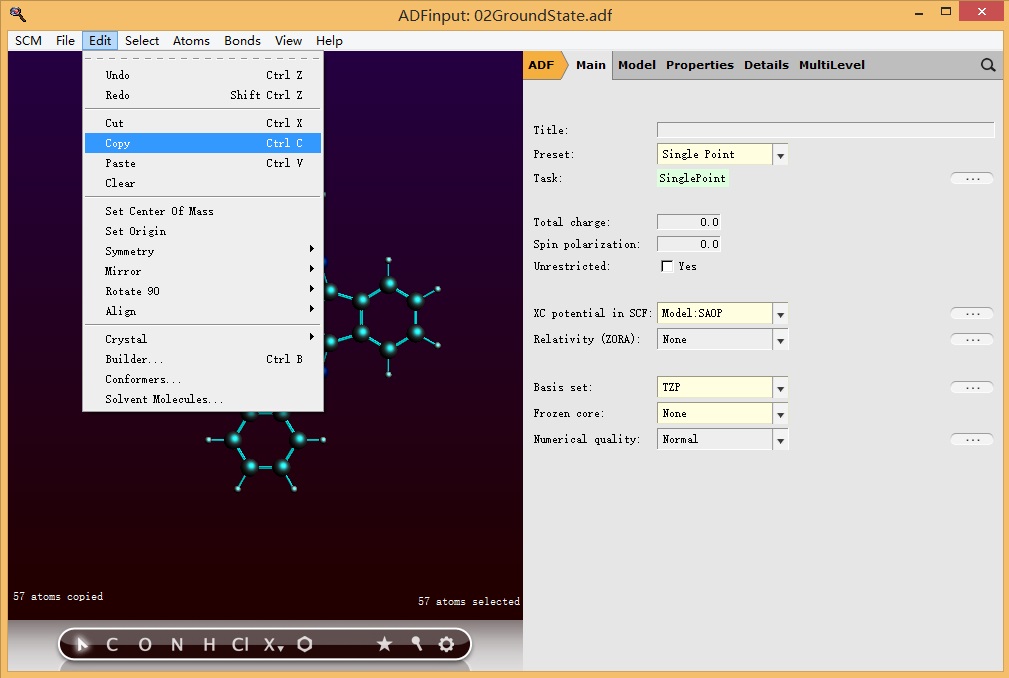
然后在新的ADFinput界面粘贴(或者按快捷键CTRL V),主面板参数设置如下图所示:
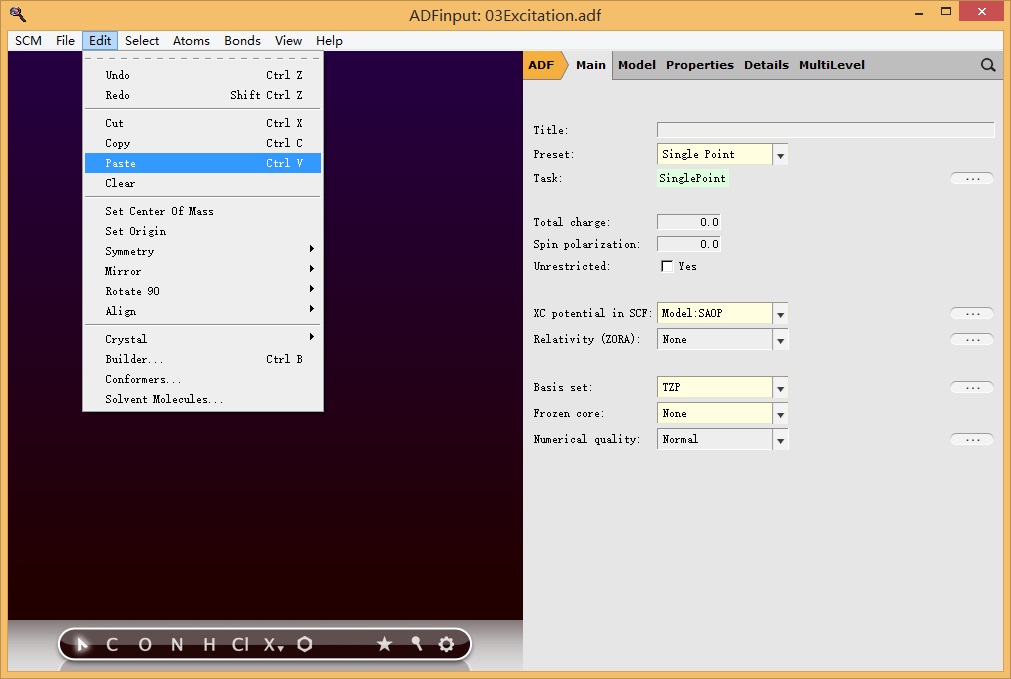
另外需要在Properties-Excitation(UV/Vis),CD设置激发态计算的参数:
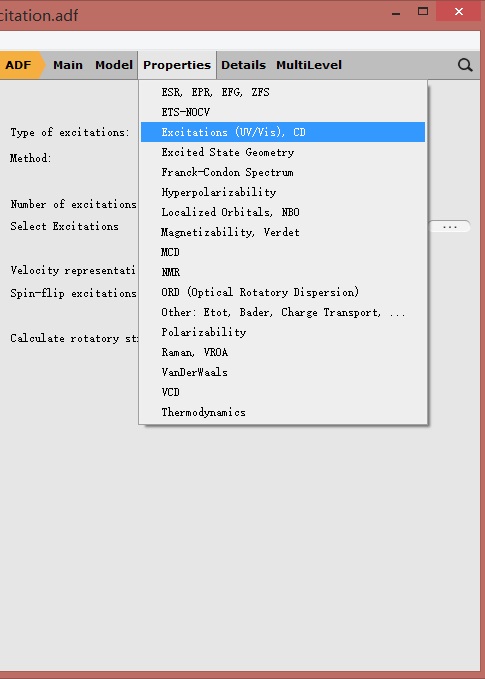
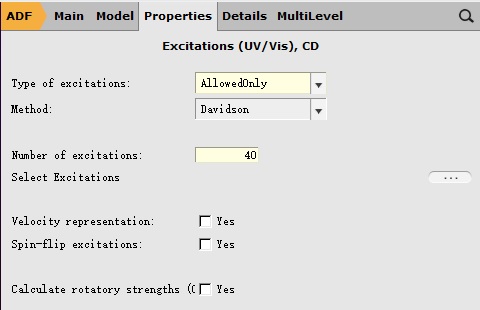
如上图所示,对于激发态的计算,一般而言只需要设置两个参数:Type of excitations和Number of excitations。
前者用于设置激发的类型:对于紫外可见吸收,基态为单重态(S0态),激发态仍然为单重态(Sn态);对于此例,选择AllowOnly与选择SingletOnly是等价的。
后者用于设置需要计算的激发态的个数:例如此例中设置为40,表示希望得到S1、S2……S40等40个激发态。理论上说,这个数值越大,计算越精确,但内存的需求也急剧增长。一个比较好的权衡,就是设置为40~60左右。
设置完毕,保存任务,例如名为03Excitation。
如果需要计算其他的激发,例如当我们希望计算原子内层轨道电子对X射线的吸收,这时候,可以使用Select Excitations,将需要激发的态指定为内层原子轨道。指定方式非常灵活,可以根据能量也可以根据轨道序号。内层轨道能量或者轨道的序号,从该结构对应的基态计算结果中,可以查找出来,进而进行指定。注意能量单位为Hartree,这是很常用的能量单位,与eV的换算关系:1Hatree=27.2113845eV。
输出结果
提交并行计算任务,计算结束之后,得到03Excitation.t21、03Excitation.out、03Excitation.logfile。
这三个输出文件是最重要的输出文件,也是最常用的输出文件,其他输出文件则一般很少用到。 查看吸收谱:

显示吸收谱如下:
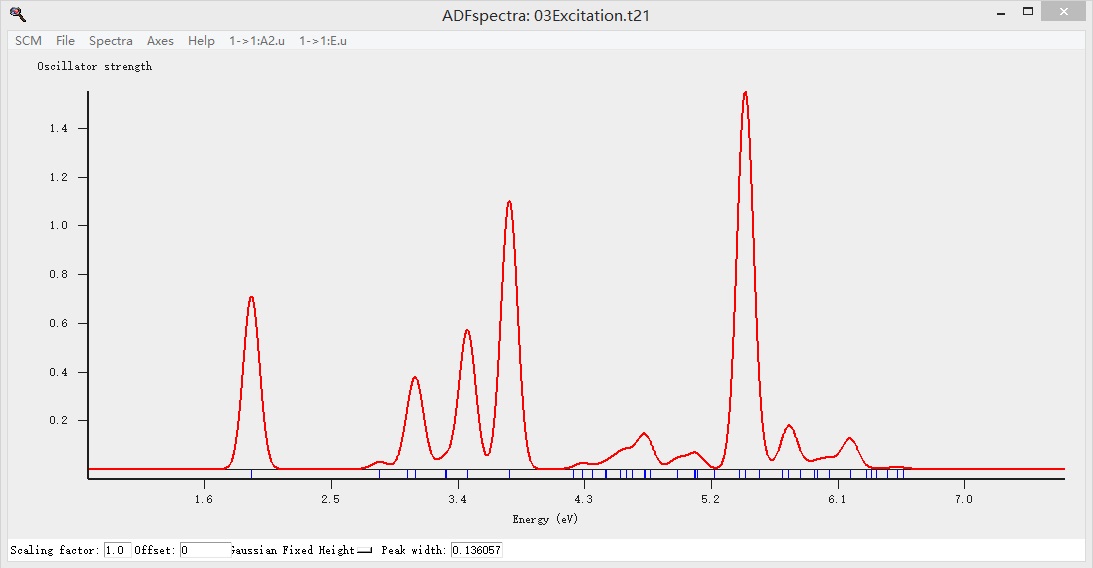
SCM LOGO > Spectra可以显示所有的谱,因为当前计算的是吸收谱(或者说激发态),因此Spectra自动显示吸收谱,如果计算的是其他的谱,就会自动显示其他谱,如果同时计算了多种谱,可以通过该窗口中的“Spectra”下拉菜单,在不同的谱中切换:
激发态分析
将鼠标置于吸收峰处,或者至于该吸收峰对应的横轴的细蓝线处,即显示该激发态或吸收峰的构成,例如下图(ADF2016以后的版本,这些信息显示在下方的一个列表中):

为能量最低的激发态(S1),其构成为:91.8%从2a1.u激发到7e.g,即从a1.u不可约表示的第2个态激发到e.g这个不可约表示的第七个轨道,而这两个态。
在SCM LOGO > levels中查看,实际上就是HOMO(最高占据轨道)和LUMO(最低空轨道),当鼠标移动到对应的能级上,即分别显示对应的轨道序号和组成:
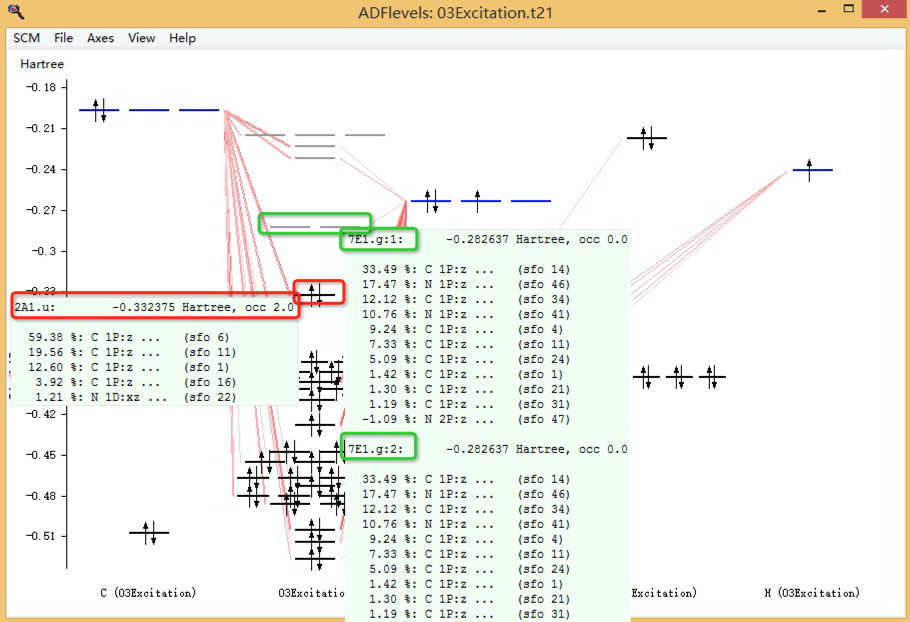
需要说明的是,上图中7 E1.g(7 E1.g也就是激发态构造图里面的7 e.g,类似的2A1.u就是激发态构造图里面的2a1.u。)是二重简并的不可约表示(因此有7 E1.g:1和7 E1.g:2两个简并轨道),所以每个E1.g能级其实都有两个轨道,能量相同(这两个能级假如要往上面填充电子的话,就可以填4个电子)。这在上图中,也可以看到。是绿色框中所示的两个轨道。
同时该激发态的振子强度为0.7097.振子强度与吸收强度成正比。则文献的列表中对应着f的那一列,例如对于S1态振子强度为0.7356。
对应地,我们在文献中看到作者的计算结果中激发态的列表:

能量最低的激发态,也就是表中的第一个激发态。其中state列,列举的是激发态的名称,字母例如Eu表示激发的不可约表示,字母左上角的数字表示激发态的自旋多重度,字母前面的数字则是标记激发态在该不可约表示中的排序,数字为1表示该不可约表示的激发的最低激发态。
上表中第一个激发态,激发能最低,实际上就是S1态。构成中,有92%是从HOMO激发到LUMO,其他小的成分则略有差别。我们一般只关心主要组分。其他小组分可靠性并不高。
需要说明的是,我们没有使用冻芯近似,因此基态轨道的不可约表示中,轨道的排序与文献中略有不同,例如此例中,我们的5a2u对应着文献中的4a2u。这种对应关系,可以从levels中找出来,HOMO往下a2u轨道分别为6a2u、5a2u……而文献中HOMO往下,则分别为5a2u、4a2u……,对应关系即因此而找到。
以上是激发态的计算完毕。
需要说明的地方:
1)通常而言,激发能与基态计算得到的轨道能关系很大,激发能实际上可以认为由两项构成:
第一项,即占据轨道和空轨道之间的能级差。拿此例S1态来说,也就是HOMO和LUMO能级之差。这一项往往占激发能的大部分,甚至达到90%。因此很多时候,我们用HOMO、LUMO能级直接来估算最低吸收峰。但也需要注意,有的时候,例如HOMO-LUMO为禁阻跃迁,那么最低激发就不是HOMO-LUMO激发,这种估算也就无效。
第二项,可以认为是占据轨道和空轨道之间的“耦合作用”,这种耦合作用大部分情况下很弱,但有时候很强,轨道的对称性对这种耦合影响很大。当耦合项很大的时候,前面提到的估算吸收峰的方式就不可靠了。
有的时候一个激发态的主要组分是好几个跃迁,这个时候,这种估算方式就更加无效了。
2)基于前面的原因,不同的泛函对激发能的影响也就非常大。因为不同的泛函计算得到的能级差往往差别很大。一般性地说,LDA计算得到的能级差最小,其次是PBE等,再次是B3LYP等杂化泛函或者SAOP。另外,冻芯与否,对能级也有较大的影响,但对能级差的影响次之。因此,一般而言,在激发态的计算时,取消冻芯近似(即:使用全电子基组)是恰当的。
3)激发态的质量:一般而言,例如B3LYP对于有机体系的低激发态,可靠性往往都是不错的,但更高的激发态,可靠性则会变差,越高的激发态,可靠性越差。其主要原因,往往也在于DFT方法本身,对于较高的空轨道能量、较内层的占据轨道能量的计算,效果都比较差。越是离HOMO-LUMO远的轨道,能量差的越多。更深层次的原因则是:DFT原则上,只有HOMO、LUMO的能级与真实的IP、EA有对应关系,而其他能级实际上与电子能级并没有对应关系;我们把DFT能级当作电子能级来使用,实际上是一种很粗糙的近似。
4)一般主要从吸收谱中波长较长的部分进行对照,波长较短的部分,跟我们计算的激发态的个数有关(默认计算10个激发态)。如果只计算少量激发态,实际上得到的吸收峰丢失了大量的短波部分的信息。