Ag-Si界面的肖特基势垒和电输运性质
版本:2016.1
在现代技术中,金属和掺杂半导体的接触无处不在,在很多重要领域中都有应用,比如太阳能光伏。ATK 配置了一个有助于研究这种界面的工具箱,因为它能够正确地解释半导体的带隙和掺杂,并且采用在物理上准确的边界条件描述界面。
这个教学案例演示如何运用 ATK 和 NEGF (非平衡格林函数)方法计算和分析金属-半导体界面特性。因此,我们将重现近日出版的 “General atomistic approach for modeling metal–semiconductor interfaces using density functional theory and non-equilibrium Green’s function” [cSMB+16] 一文中所报道的主要计算和理论分析部分,以 Ag-Si 界面作为例子参考。但请您注意,这里介绍的方法是完全 通用的,可以应用于任何金属-半导体界面。
您将按照以下步骤指南操作:
- 构建器件
- 计算并画出投影局域态密度
- 计算理想因子
- 估算、分析肖特基势垒
- 计算谱电流
提示
本教程使用特定版本的QuantumATK创建,因此涉及的截图和脚本参数可能与您实际使用的版本略有区别,请在学习时务必注意。
注意
QuantumWise 的案例研究主要针对的是可以熟练使用 VNL 和 ATK 的用户。特意设计简洁的软件指令以便用户更专注于科学研究。
关于 VNL 和 ATK 使用方法的更多详细基本信息,可以参考教程 Introduction to VNL和 Transport calculations with ATK。

构建器件
提示
如果您想跳过这个部分,只需要下载一个器件构形作为 ATK 的 Python 脚本: ↓device.py。
第一步,弛豫 Ag 和 Si 的块体结构。用 ![]() Builder 的数据库创建结构,并依次发送到
Builder 的数据库创建结构,并依次发送到 ![]() Scripter。在这两个脚本中都添加一个参照以下设置的
Scripter。在这两个脚本中都添加一个参照以下设置的 ![]() New Calculator 模块:
New Calculator 模块:
- ATK-DFT
- LDA 交换关联函数
- 11×11×11 的 k 点取样
添加 ![]() OptimizeGeometry 模块,分别设置 force tolerance 和 stress tolerance 为 0.01 eV/Å 和 0.001 eV/Å3,不勾选 Constrain cell 选项框。运行这两个脚本,得到优化后的结构,该计算可以很轻易地在您的本机上实现。
OptimizeGeometry 模块,分别设置 force tolerance 和 stress tolerance 为 0.01 eV/Å 和 0.001 eV/Å3,不勾选 Constrain cell 选项框。运行这两个脚本,得到优化后的结构,该计算可以很轻易地在您的本机上实现。
现在用 Builder 里的 Surface(Cleave) 工具创建 Ag 和 Si 的 (100) 面,并运用正如在教程 Building an interface between Ag(100) and Au(111) 中描述的方法在此生成界面。结构中应该包含有 6 层 Ag 和 9 层 Si。在 Select Surface Cells 窗口, Straining method 下面选 Strain first surface,然后选择如下图所示的单元。

提示
您可以在 The Interface Builder in VNL里学习到更多 Interface Builder 的技术注解。
界面结构的初始猜想
用 Repeat 工具沿B方向重复该界面2次。接下来,先用 Stretch Cell 工具沿 C 向量拉伸 30%,然后用 Center 工具将原子在 C 方向居中,从而在结构周围增加些空间。
在 Ag-Si 界面,Si 悬空键最初被放置在 Ag(100) 面中空位。为了找到最稳定的界面结构,您应该也研究下顶位和桥位的情形。
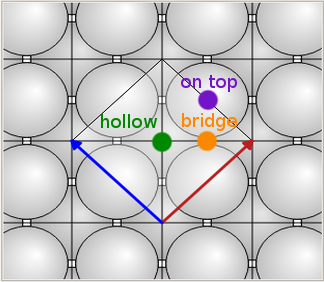
选定所有的硅原子,打开 Move ![]() 工具,通过双击选择最左边顶端的硅原子作为锚点。在 Move 窗口中增加 c 值使 Si(100) 面距 Ag(100) 面距离为 4 Å,然后将结构重命名为
工具,通过双击选择最左边顶端的硅原子作为锚点。在 Move 窗口中增加 c 值使 Si(100) 面距 Ag(100) 面距离为 4 Å,然后将结构重命名为 interface_hollow。复制该结构,再次使用 Move ![]() 工具。这次使用 Fractional 选项,输入 a = 0.5,b = 0.75(如下图),给该结构命名为
工具。这次使用 Fractional 选项,输入 a = 0.5,b = 0.75(如下图),给该结构命名为 interface_ontop。最后,再次复制结构,设置 a = 0.25,b = 0.625,命名为 interfaca_bridge。

注意
一般情况下重复结构不是必要的,在这里我们这样做是为了获得和参考文献 [cSMB+16] 中相同的结构模型。
电极弛豫
接下来,您应该在 z 方向上弛豫 Ag 电极。在创建界面时已经在 x 和 y 方向上给电极施加了应力,因此可以根据材料的泊松比对电极在 z 方向上弛豫。您可以通过先创建一个器件构形获得电极(给以上生成的界面中的其中一个应用 Device From Bulk 插件),然后用 ![]() 图标将其分离。使用 ATK-DFT 计算器,选择 LDA 和 6×3×1 k 点网格,force tolerance 和 stress tolerance 跟之前对块体弛豫时的设置相同。计算后您会发现电极 C 向量的长度增长了 9.57 %。为了保持一致,您应该在三个界面构形 Ag 的部分应用相应的延伸率。再次运用 Stretch Cell 工具:选中所有 Ag 原子,沿 C 向量方向的拉伸输入 1.0957,然后选择 Selected atoms are stretched。
图标将其分离。使用 ATK-DFT 计算器,选择 LDA 和 6×3×1 k 点网格,force tolerance 和 stress tolerance 跟之前对块体弛豫时的设置相同。计算后您会发现电极 C 向量的长度增长了 9.57 %。为了保持一致,您应该在三个界面构形 Ag 的部分应用相应的延伸率。再次运用 Stretch Cell 工具:选中所有 Ag 原子,沿 C 向量方向的拉伸输入 1.0957,然后选择 Selected atoms are stretched。
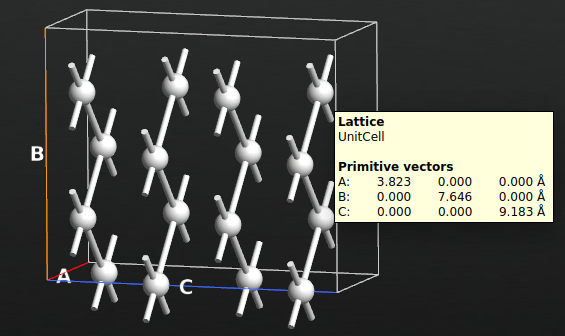
中心区域弛豫
器件构形弛豫的常规步骤已在在教程 Advanced device relaxation 中做了详细说明。为了确定这三个界面结构中哪一个是最稳定的,您需要先把它们当作块体结构(也就是弛豫器件的中心区域)进行优化。
首先,用 Lattice Parameters 插件给 3 个界面结构在 C 向量方向拉伸 30% 以增加额外空间。然后用 Center 工具使构形沿 C 方向居中。最后,用 ![]() Hydrogen passivation 工具钝化处理离界面最远的 Si(100) 面上的悬空键。需要注意的是,这个操作可能会给硅部分的左右侧增加 H 原子。而显然置于界面处的氢都应该被清除掉。
Hydrogen passivation 工具钝化处理离界面最远的 Si(100) 面上的悬空键。需要注意的是,这个操作可能会给硅部分的左右侧增加 H 原子。而显然置于界面处的氢都应该被清除掉。
现在,您已经准备好要弛豫每个构形了。计算器的设置跟弛豫 Ag 电极时的相同,在 ![]() OptimizeGeometry 模块中按照如下选用参数:
OptimizeGeometry 模块中按照如下选用参数:
- Force tolerance 为 0.02 eV/Å。
- 不要使应力最小化。
- 用 Add Constraints 小程序给下图显示的 Ag 原子施加 Fixed 约束,Si(和 H)原子施加 Rigid 约束。
最后,在脚本中添加 ![]() TotalEnerg y 分析模块。
TotalEnerg y 分析模块。

运行计算,这个过程需求略高,如果用 1 个 16 核的节点大概需要 20 小时。您会发现那些硅连接着 Ag(100) 面中空位置的结构是最稳定的(具有最低的总能量,如下图)。接下来,所使用的都是这个含有最低能量的构形。
| Total Energy(eV) | |
|---|---|
| Hollow | -76536.37 |
| On top | -76535.46 |
| Bridge | -76535.40 |
器件弛豫
从优化后 “hollow” 界面结构中移出钝化的氢原子,用 Device From Bulk 插件生成一个器件。除了沿 C 方向上的 k 点取样必须增加到 401( 6×3×401 网格)外,最终器件弛豫所用计算器和结构优化参数跟上述块体弛豫时完全相同。
用 16 核的话计算需要 2 小时左右。如果您不打算运行计算,可以在此处下载弛豫后的器件构形 ↓device_hollow.py。

调试 MGGA c 参数
我们使用 TB09 meta-GGA 泛函计算器件的投影局域态密度和透射谱。为了准确描述界面的硅所在侧,[TB09] c 参数要匹配硅的带隙。更详细的步骤信息可以参考教程 Meta-GGA and 2D confined InAs。计算时间短,可在本机实现。
参照以下步骤:
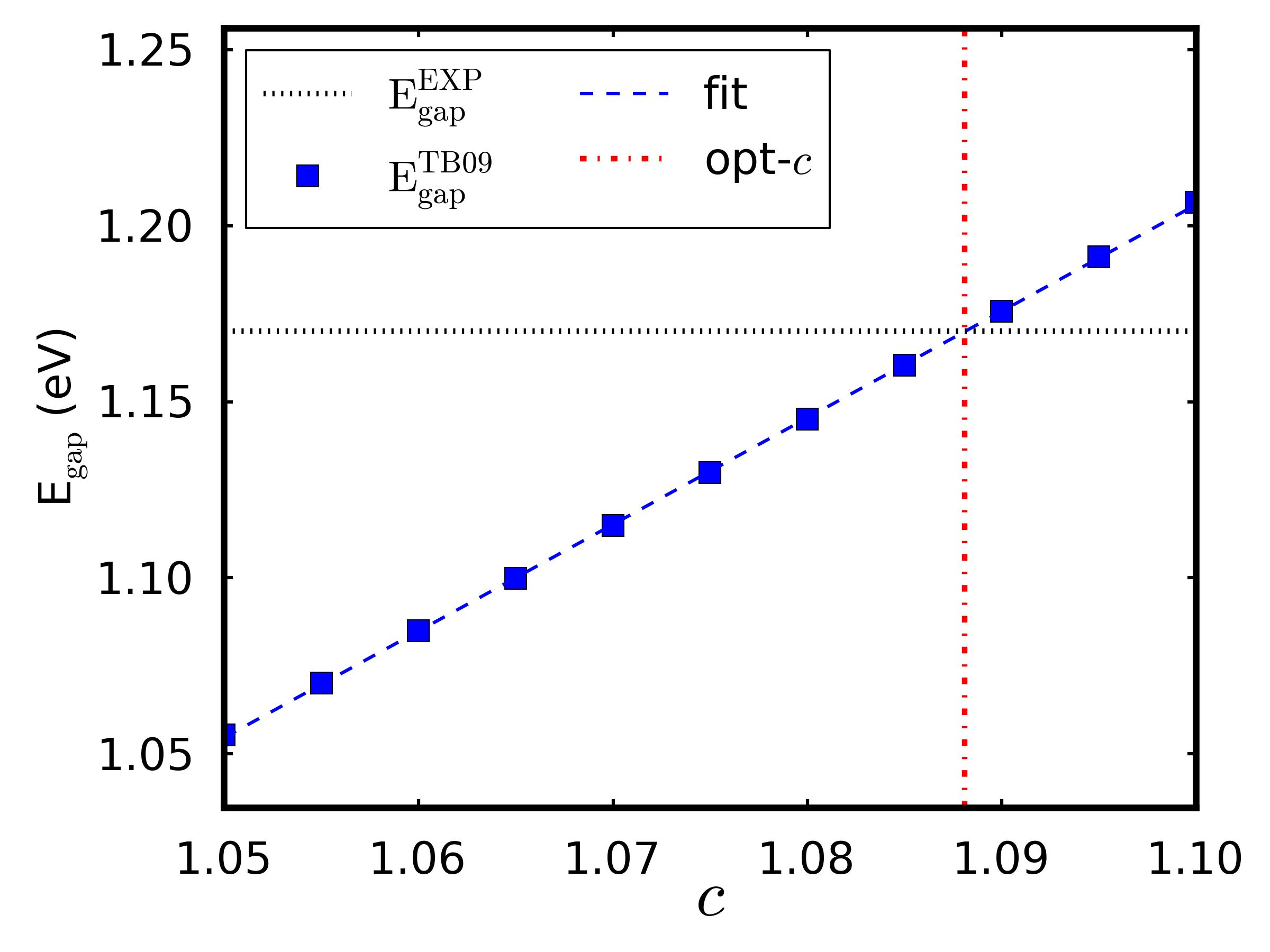
1. 计算优化后硅块体的能带结构,采用 ATK-DFT 的 MGGA 泛函和 11×11×11 k 点。从结果可得到带隙为 1.16 eV和自洽确定的 c 参数为 1.0841。
2. 做固定 c 值约为 1.0841 的类似计算。这步可以通过在一个 ATK 的 Python 脚本中循环不同的 c 值实现。采用 ![]() Editor 输入如下循环:
Editor 输入如下循环:
for tb09 in (1.05,1.055,1.06,1.065,1.07,1.075,1.08,1.085,1.09,1.095,1.1): # ------------------------------------------------------------- # Calculator # ------------------------------------------------------------- #---------------------------------------- # Exchange-Correlation #---------------------------------------- exchange_correlation = MGGA.TB09LDA(c=tb09)
谨记 Bandstructure 模块应该被包含在该循环中,放在 Calculator 部分的下方。用 nlsave(‘Si_bulk_fitc.nc’, bandstructure) 将所有的能带结构保存在一个单独的数据文件中。
3. 用脚本 ↓c_fit.py 拟合实验带隙 1.17 eV [cKit04] 的 c 值。需要 3 个输入参数:用过的 c 值列表(tb09),包含能带结构计算的 .nc 文件(fname),实验带隙(sk)。
最佳的 c 值应该在 1.0881 附近。
硅掺杂和耗尽层长度
现在已经做好准备给器件的硅部分掺杂。在 ![]() Builder 中打开弛豫过的器件构形,选中所有的 Si 原子,用 Doping 插件对右电极施加浓度为 1019 e/cm3 的 n 型掺杂。
Builder 中打开弛豫过的器件构形,选中所有的 Si 原子,用 Doping 插件对右电极施加浓度为 1019 e/cm3 的 n 型掺杂。
考虑器件中耗尽层的长度事很重要的。器件需要比半导体的屏蔽长度更长,以便硅电极边界处硅侧的电势与块体硅的相近。这可以通过研究 Hartree 势随硅中心区域长度变化收敛得到。至于一个好的初始猜想,您可以参考一些典型金属-半导体界面的耗尽层长度 [cKP05];1019 cm-3 的掺杂相对应于 100 Å 左右的耗尽层长度。
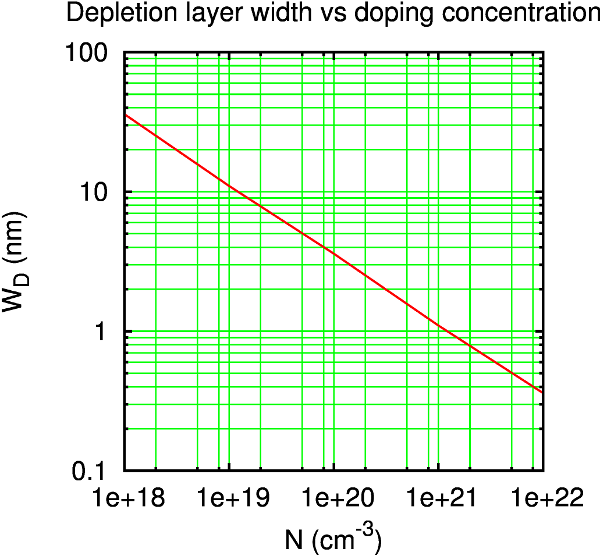
提示
由于金属的屏蔽长度要短得多,只需要收敛界面硅侧的长度。Ag 的 6 个原子层就完全足够了。
注意
ATK 在 2015 版本和其之前的版本中, ![]() HartreeDifferencePotential 分析对象叫做 ElectrostaticDifferencePotential。
HartreeDifferencePotential 分析对象叫做 ElectrostaticDifferencePotential。
使用 Central Region Size 插件增加中心区域硅侧的长度。设置长度分别为 80 Å、100 Å、120 Å 和 140 Å,将每个器件构形发送给 ![]() Scripter。采用与器件弛豫时相同参数设置的计算器,添加
Scripter。采用与器件弛豫时相同参数设置的计算器,添加 ![]() HartreeDifferencePotential 模块。
HartreeDifferencePotential 模块。
注意,随着长度的增加,计算收敛的难度也逐渐增大。在大于等于 120 Å 时,需要在计算器中调整 Iteration Control Settings:在 Scripter 窗口,更改 Script detail 为 Show defaults,发送脚本 ![]() Editor。在 Iteration Control Settings 模块,修改器件计算中需要用到的
Editor。在 Iteration Control Settings 模块,修改器件计算中需要用到的 damping_factor, number_of_history_steps 和 max_steps。
device_iteration_control_parameters = IterationControlParameters( damping_factor=0.05, linear_dependence_threshold=0.0, algorithm=PulayMixer(), preconditioner=Preconditioner.Off, start_mixing_after_step=0, number_of_history_steps=12, max_steps=400, tolerance=0.0001, mixing_variable=HamiltonianVariable, )
最大的器件用 16 个核计算需要 13 个小时。当所有的计算结束后,结果显示在 LabFloor,您可以用 1D projector 分析工具画出电势沿 Z 方向的变化图。硅电极边界附近的电势应该是很平坦的。
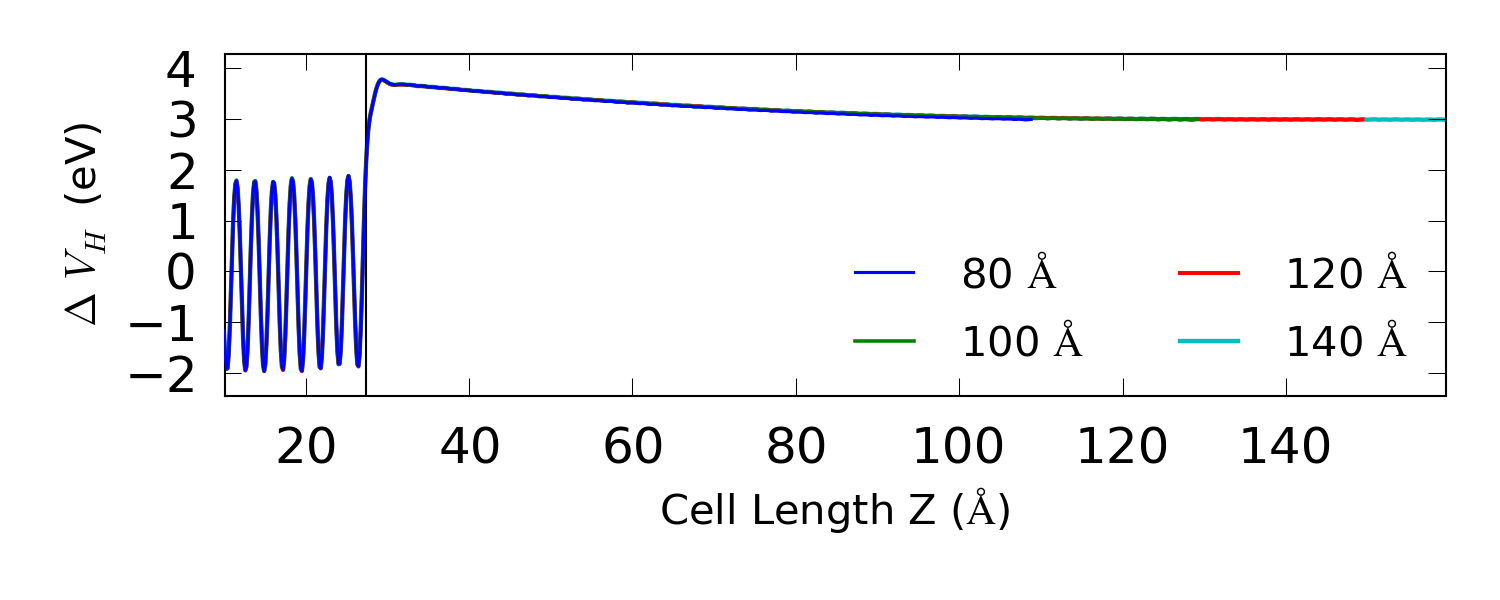
电势的摆动使它很难判断是否已经收敛到电极值。电势的 macroscopic average$\langle \Delta V_H \rangle$ 使浮动变得平滑。用脚本 ↓hdp.py 计算 Hartree 电势沿z方向的宏观平均值 [cBBR88]。这个脚本还需要再编译,因为它包含很多界面结构的特定输入参数。
| gauss_left: | 左侧电极材料相连两个原子层沿 Z 方向的距离(单位为 Å)。 |
| gauss_right: | 同上,但是对象是右侧电极材料。 |
| zcoord_int: | 界面位置即左侧材料的最后一个原子和右侧材料的第一个原子的中点的Z坐标(单位为 Å)。 |
| fname: | 包含平均电势的 .dat 输出文件名称。 |
编译好这些参数后,每次运行一个包含着不同构形 HartreeDifferencePotential 分析的 .nc 文件脚本。
atkpython hdp.py device.nc
这里 device.nc 文件应该更改为实际的 .nc 文件名。
您可以用脚本 ↓hdp_plot.py 比较产生的 .dat 输出文件。修改一下输入参数:
| fnames: | .dat 文件名称 |
| names: | 硅中心区域的长度 |
| Zcoord_int: | 同上说明(↓hdp.py 的输入) |
编译脚本后运行,生成下图:
atkpython hdp_plot.py
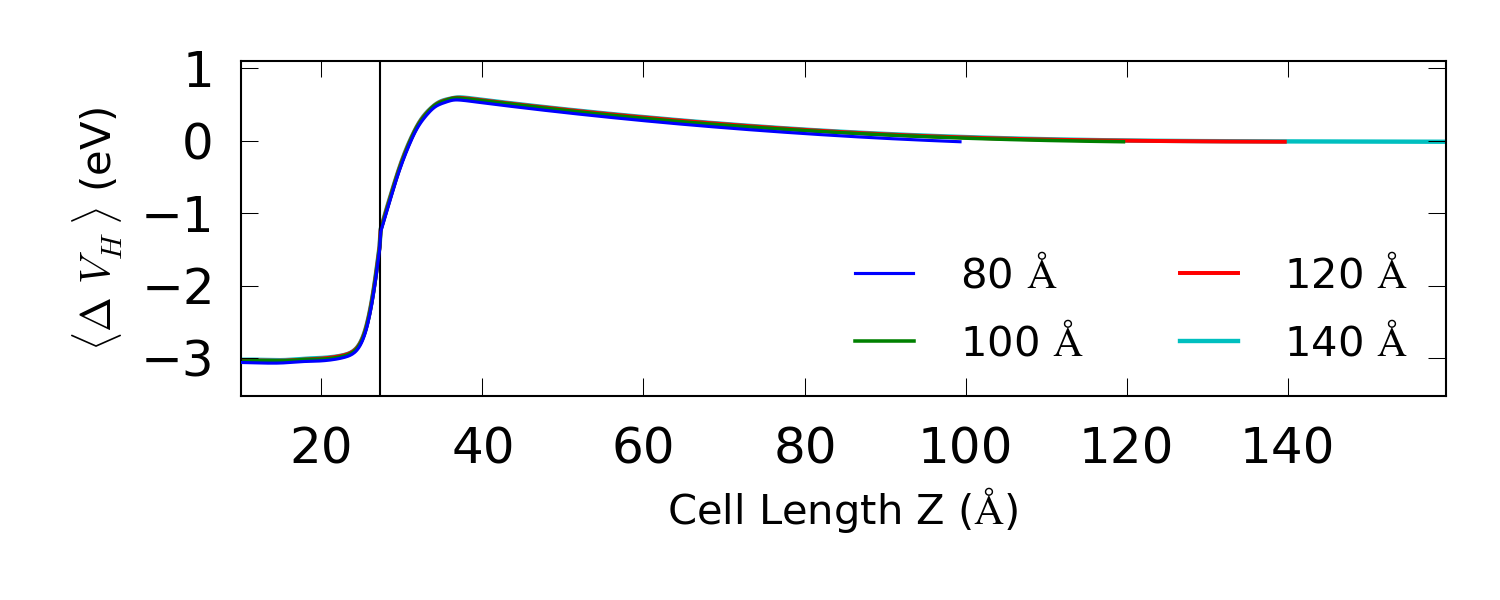
我们可以从图中得出结论:120 Å 的硅中心区域长度是足够的。注意,耗尽层的长度可能会随着偏压发生轻微的变化。现在,您就有了最终的 ↓ device.py。
注意
上图中界面边缘的扭折是由平均化程序引起的,此处不同宽度的高斯函数在 Ag-Si 边界是相匹配的。
投影局域态密度
投影局域态密度(PLDOS)提供了非常有用的界面能带图可视化功能。在 ![]() Scripter 的
Scripter 的 ![]() Analysis from File 模块下载已保存的器件构形和附加的收敛计算器。然后添加
Analysis from File 模块下载已保存的器件构形和附加的收敛计算器。然后添加 ![]() ProjectedLocalDensityOfStates 模块,设置为:能量范围为 -2 eV 到 +2 eV 间的 401 个能量点,18×9 的 k 点网格。在下载构形位置处的 .nc 文件中保存结果。
ProjectedLocalDensityOfStates 模块,设置为:能量范围为 -2 eV 到 +2 eV 间的 401 个能量点,18×9 的 k 点网格。在下载构形位置处的 .nc 文件中保存结果。
这个计算用带有 64 G 内存的 16 个核大概需要 7 小时。计算结束后,结果显示在 LabFloor,您可以用 Projected Local Density Of States 分析插件画出沿构形输运方向的 PLDOS:
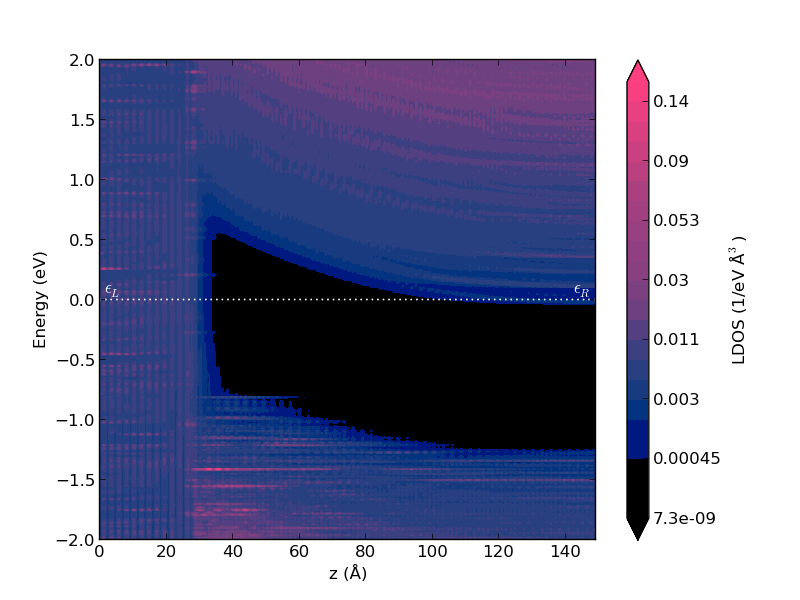
您还可以用脚本 ↓ pldos_hdp.py 绘制平均 Hartree 差异势图和 PLDOS图。它需要包含 HartreeDifferencePotential 和 ProjectedLocalDensityOfStates 分析项目的 .nc 文件,与 ↓ hdp.py 中相同的输入参数以及一些绘图的参数设置:
| leftname: | 左侧界面材料的名称 |
| rightname: | 右侧界面材料的名称 |
| lenleft: | 图中所示左侧材料的数目(单位为 Å) |
使用生成的 .nc 文件以 atkpython pldos_hdp.py device.nc 运行脚本。
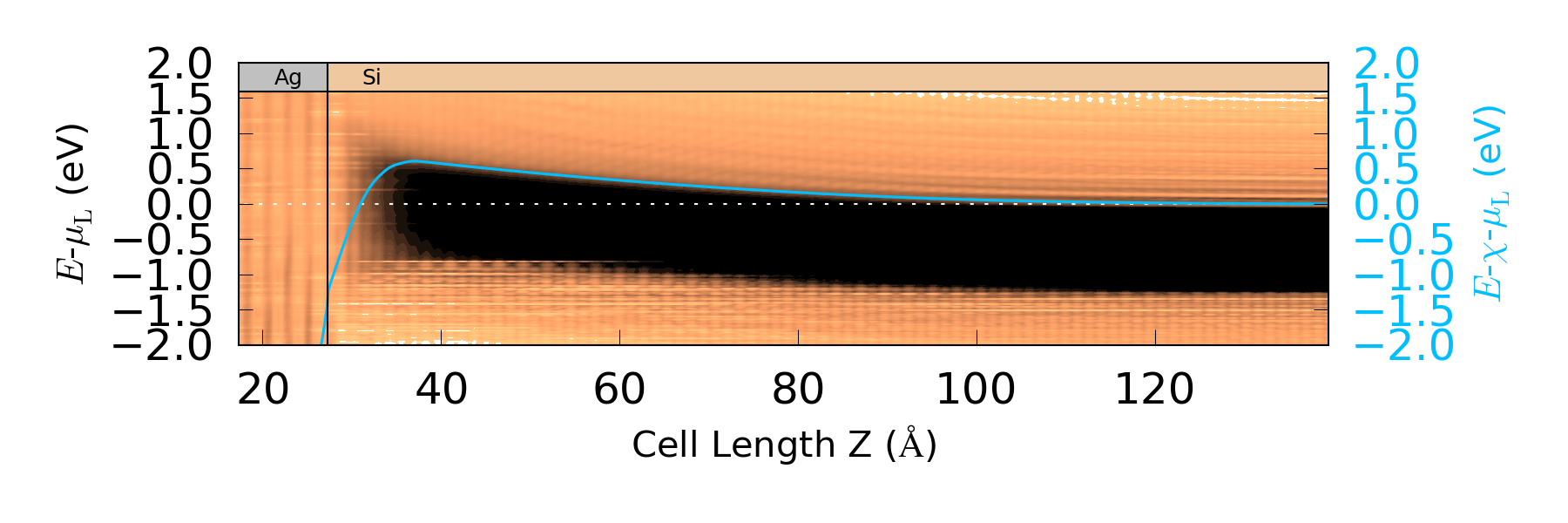
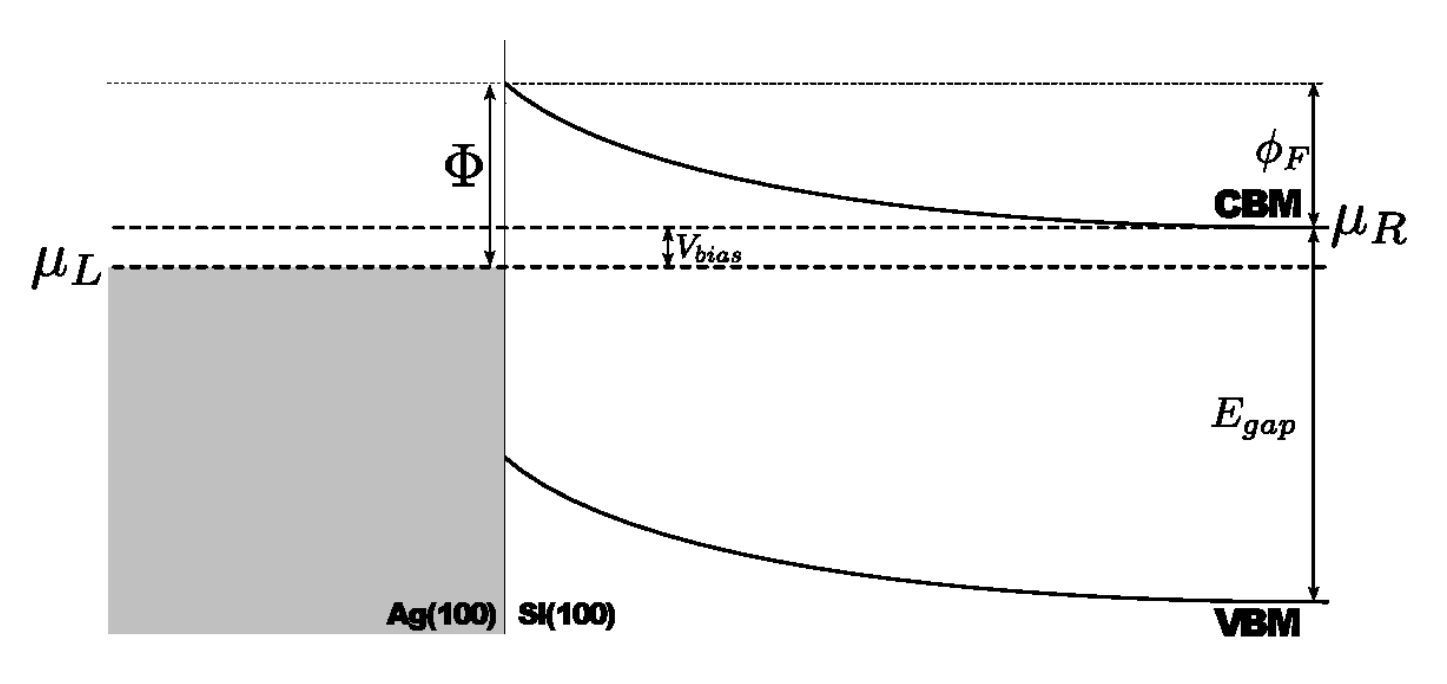
您可以很容易地从上图中看到界面边缘右侧的金属诱导带隙态。而且平均电势在界面处及更远处都很好地跟随着导带的最小能级。因此,我们可以通过计算电极的化学势 $\mu_L$ 和 $\langle \Delta V_H \rangle$ 最大值的差别来估算肖特基势垒 $\Phi^{pot}$。绘图的脚本会将该值和图一起呈现出来。您将得到一个大约为 0.606 eV 的势垒,与实验结果一致,可参考 [cBML13] 和 [cGASL10]。该脚本还能输出随 $\mu_L$ 变化对应处的导带最小值位置,在稍后分析谱电流时会需要这个值。
有限偏压计算
接下来我们将研究 I-V 特性,需要用到 ![]() Analysis from File 功能和
Analysis from File 功能和 ![]() IVCurve 分析模块。为分析选择以下设置:
IVCurve 分析模块。为分析选择以下设置:
- 15 个点,偏压范围为 -0.3 到 +0.3 V
- 401 个点,能量范围为 -2 到 +2 eV
- 18×9 的 k 点网格
采用 16 个核的话计算大约需要 15 小时。您可以使用 IV-Plot 插件画出计算电流和偏压的曲线,或是用脚本 ↓ IV.py 绘出电流强度和偏压的图。脚本中需要包含器件构形和 I-V 曲线项的 .nc 文件。

这个界面显示出了类二极管行为,电流在正向偏压下急速增加,在负向偏压下增速较平缓。
注意
计算模拟得到的电流要明显低于在出版文章 [cSMB+16] 中报道过的数据。相关解释请参照 Note on the variation of the current 的注释。
理想因子
在参考资料 [cSK06] 中,热离子发射理论描述肖特基二极管的 I-V 特性:
$$I = I_0 \mathrm{e}^{\frac{qV_{bias}}{nk_BT}} \left(1-\mathrm{e}^{-\frac{qV_{bias}}{k_BT}}\right),$$
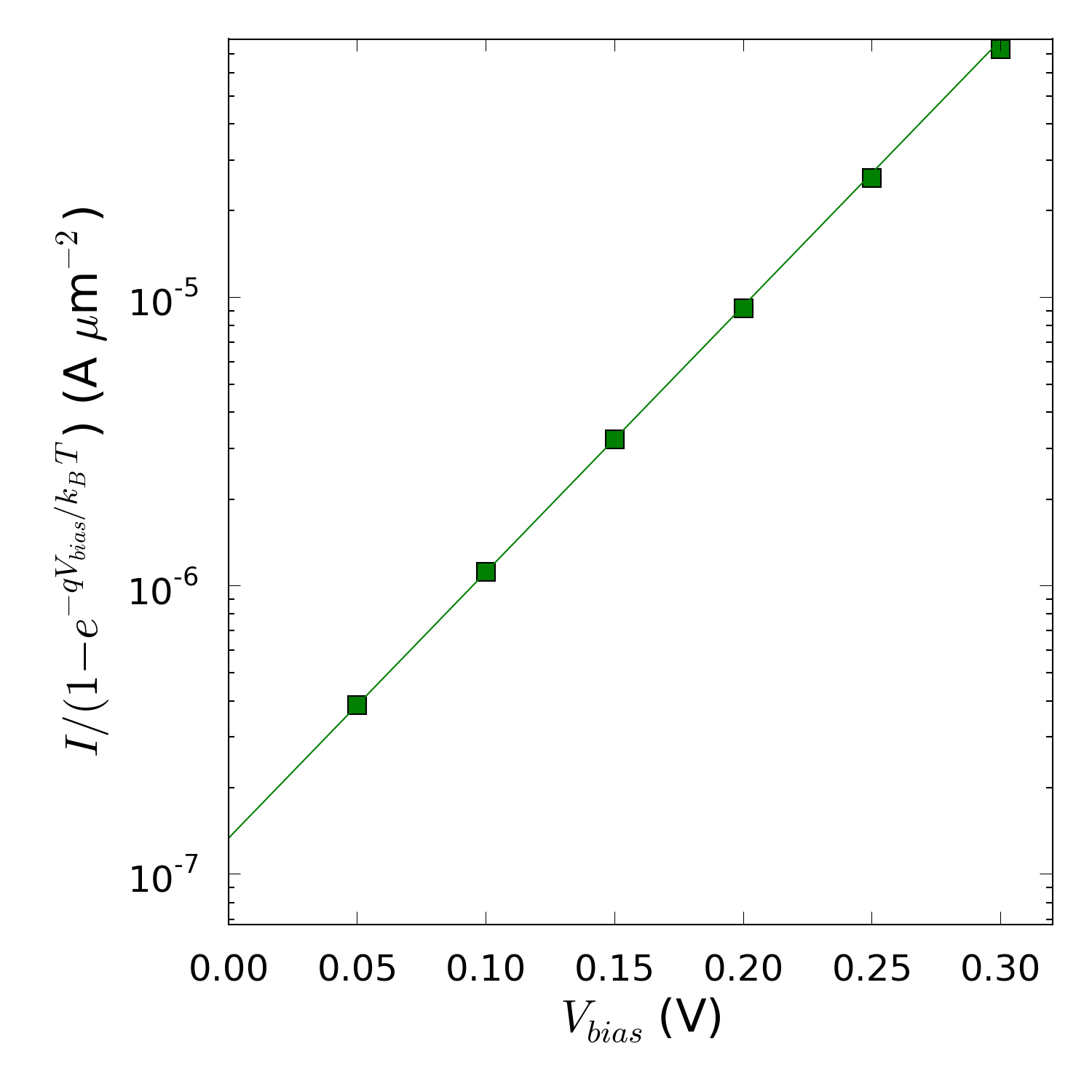
这里,$q$ 是元电荷,$k_B$ 是玻尔兹曼常数,$T$ 是温度,$I_0$ 是饱和电流,$n$ 就是所谓的理想因子。理想因子是用来衡量界面与理想状态肖特基二极管的相似程度,$n$ = 1 时代表在理想状态下。您可以从 $I/(1-\mathrm{e}^{-qV_{bias}/k_BT})$ 随 $V_{bias}$ 的变化的对数坐标图的斜率提取出理想因子的值。用脚本 ↓IV-n-log.py 做出对数坐标图并计算出理想因子。但是您需要编译一些输入参数:
| fliename: | 与 ↓IV.py 相同的 .nc 文件 |
| T: | 已经运行计算时的温度 |
| numpoints: | 用于寻找理想因子而在拟合中包含的偏压点的数目 |
运行脚本,您会得到一个 $n$ = 1.8208 的大约值,也就意味着体系明显偏离了理想状态的肖特基二极管行为。
肖特基势垒
活化能(AE)法 [cSK06] 是广泛应用在实验中提取肖特基势垒的方法。该法从 $\ln(I/T^2)$ 随 $1/T$ 变化的类 Arrhenius 曲线中得到肖特基势垒,
$$\ln(IT^2) = \ln(AA^*) - \frac{q}{k_B} \left( \Phi^{AE} - \frac{V_{bias}}{n} \right) \frac{1}{T} \Rightarrow$$
$$\phi^{AE}(V_{bias}) = \Phi^{AE} - \frac{V_{bias}}{n} = -\frac{k_B}{q} \frac{\mathrm{d}[\ln(I/ T^2)]}{\mathrm{d}(1/T)}.$$
用脚本 ↓ IvsT.py 计算在 250 K 到 400 K 的温度范围内,每一个施加正向偏移的输运。用 Landauer–Büttiker 表达式对输运进行线性响应计算,
$$I = \frac{2q}{h} \int T(E,\mu_L,\mu_R) \left[ f \left( \frac{E-\mu_L}{k_BT} \right) - f \left( \frac{E-\mu_R}{k_BT} \right) \right] \mathrm{d}E,$$
输运系数 $T(E,\mu_L,\mu_R)$ 已经在计算 IV 曲线时温度 300 K 时进行过自洽评估。
脚本需要3个输入参数文件才能运行
| fname: | 含有 IVcurve 分析的 .nc 文件 |
| Tmax: | 计算过程中的最高温度(单位为 K) |
| Tmin: | 计算过程中的最低温度(单位为 K) |
运行脚本生成的 .dat 文件包含了每个正向偏压的 I-T 特征。从这些文件,您可以用 ↓arrhenius.py 生成模拟的 Arrhenius 图。输入参数有 Tmax 和 Tmin,其他的如下所示:
| If: | 从 ↓IV-n-log.py 中得到计算的理想因子 |
| voltages: | 已模拟的计算中偏向正压(单位为 V) |
| fname: | 含有效势垒高度(单位为 eV)的输出文件名称 |
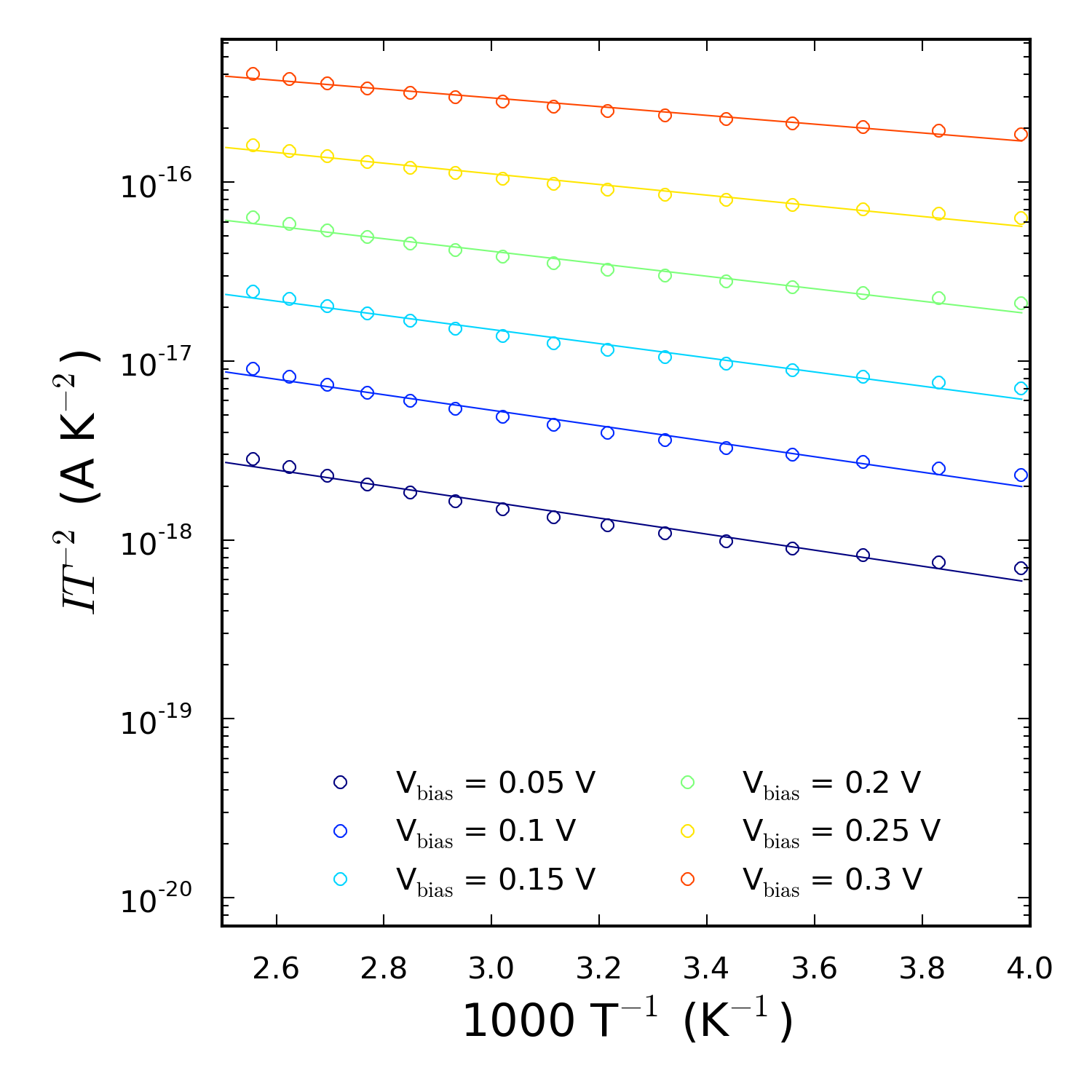
脚本运行后得到上图,还可以返回肖特基势垒高度 $\Phi^{AE}$ 和每个偏压有效势垒高度 $\phi^{AE}(V_{bias})$($\phi^{AE}$ 是从块体硅电极的导带最小值到 $\langle V_H\rangle$ 最大值的势垒)。有效势垒高度在之后的分析中以 .dat 文件形式输出。您将会发现肖特基势垒随偏压的增大而增大。这是非物理性且不应发生的,表明了 AE 法仅能对这个界面提供有限描述。
谱电流
最后的分析是对谱电流的研究,
$$I(E) = \frac{2q}{h} T(E,\mu_L,\mu_R) \left[ f \left( \frac{E-\mu_L}{k_BT} \right) - f \left( \frac{E-\mu_R}{k_BT} \right) \right].$$
为了比较最大谱电流的能量和 Hartree 势的势垒,我们需要把谱电流和平均 Hartree 电势差放在同一个图中。
要考虑到计算在不同偏压下的 Hartree 势的起点问题。做到这点则要用到 ![]() Analysis from File,下载保存在由 IV 曲线计算产生的
Analysis from File,下载保存在由 IV 曲线计算产生的 incurve_selfconsistent_configurations.nc 文件中的构形,给每个构形添加 ![]() HartreeDifferencePotential 模块。计算速度快且可在本机运行。您还可以再次使用脚本 ↓hdp.py 计算每个势的平均值。
HartreeDifferencePotential 模块。计算速度快且可在本机运行。您还可以再次使用脚本 ↓hdp.py 计算每个势的平均值。
用脚本 ↓spectral_current.py 画出的平均势能和谱电流在同一张图上。您需要编辑一些参数:
| fname: | 含有IV曲线分析的 .nc 文件 |
| Hdp_files: | 含有平均势能的 .dat 文件 |
| phipot: | 用 ↓pldos_hdp.py 计算在 0 偏压(单位为 eV)处的肖特基势垒 |
| CB_min: | 同上用 ↓pldos_hdp.py 计算块体硅电极的导带最小值随 $\mu_L$ 变化的关系 |
| fname2: | 包含与热离子发射相关势垒(单位为 eV)的输出文件名 |
| Fname3: | 包含最大谱电流的能量(单位为 eV)的输出文件名 |
运行脚本会产生下图。
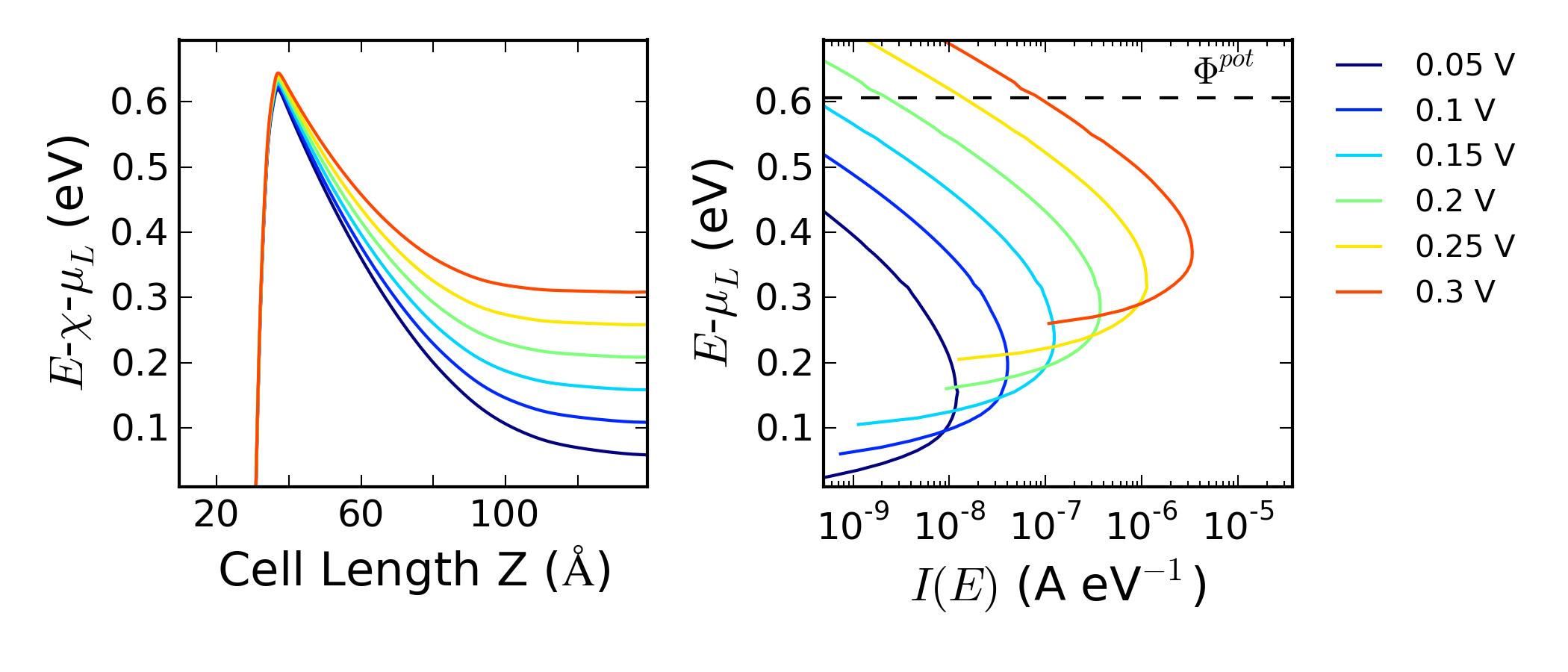
计算脚本也会返回热离子发射的估算势垒 $\phi_F$ 和如 .dat 文件中包含的每个偏压的最大谱电流能量。绘制平均势能随块体硅电子亲和能(由脚本 ↓pldos_hdp.py)和 $\mu_L$ 变化关系图以及谱电流随 $\mu_L$ 变化关系图。
器件的输运可以发生在两种不同的进程中,或是热离子发射或是穿过肖特基势垒。如果仅由热离子发射引起的输运,谱电流会在零下低于 $\Phi^{pot}$。此案例并不是上述情况。实际上对于每个偏压,最大谱电流的能量值都低于热离子发射势垒,揭示着主导输运进程的是隧道效应。
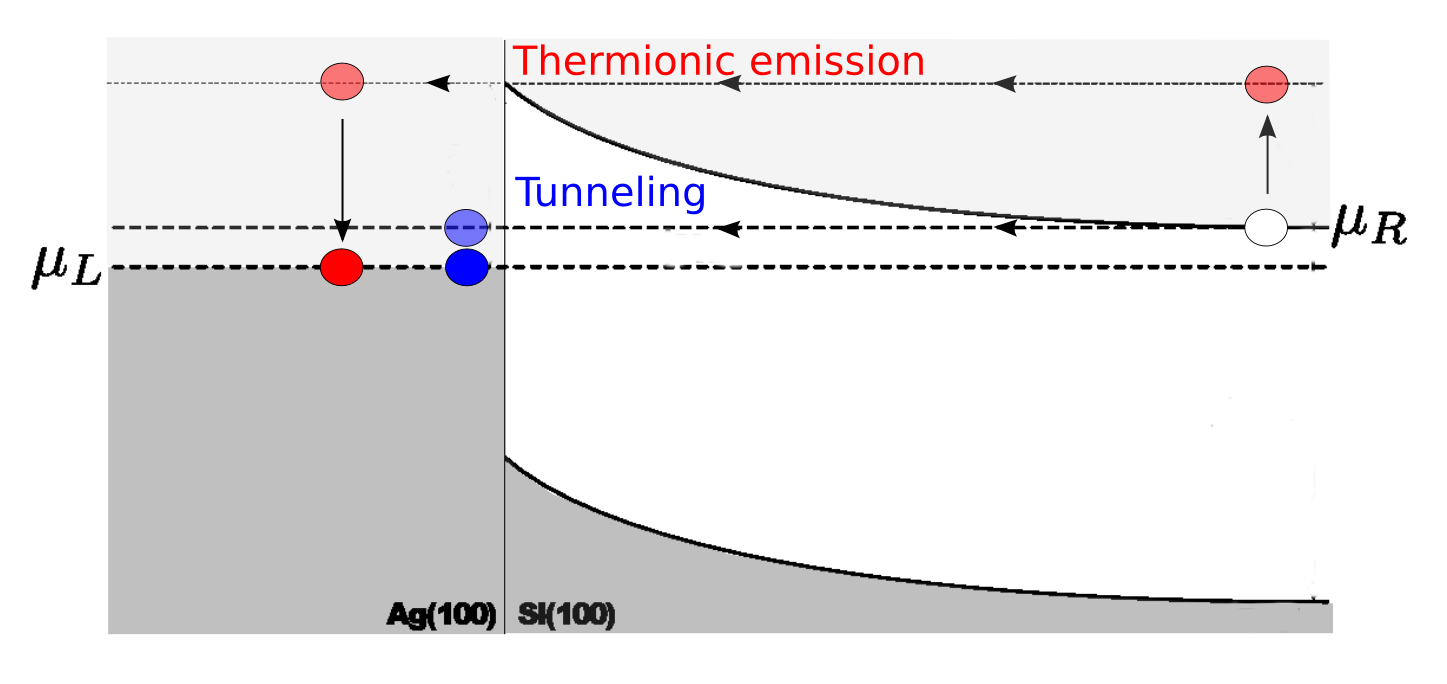
结果总结
通过绘制以下的图来对比有限偏压的结果:
- 由 ↓spectral_current.py 得到最大谱电流能量(圆圈)
- 由 ↓spectral_current.py 得到热离子发射势垒 $\phi_F$ (三角形)
- 由 ↓arrhenius.py 得到有效 AE 肖特基势垒 $\phi^{AE}(V_{bias})$ (正方形)
- $\Phi^{AE}=\Phi^{pot}$ 时,由 $\phi^{AE}(V_{bias})$ 预测到的 AE 肖特基势垒随偏压变化(虚线)
用脚本 ↓barrier_compare.py 绘图。该脚本需要 ↓arrhenius.py 和 ↓spectral_current.py 生成的 .dat 文件,↓IV-n-log.py 得到的理想因子,↓pldos_hdp.py 估算在 0 偏压处的肖特基势垒。
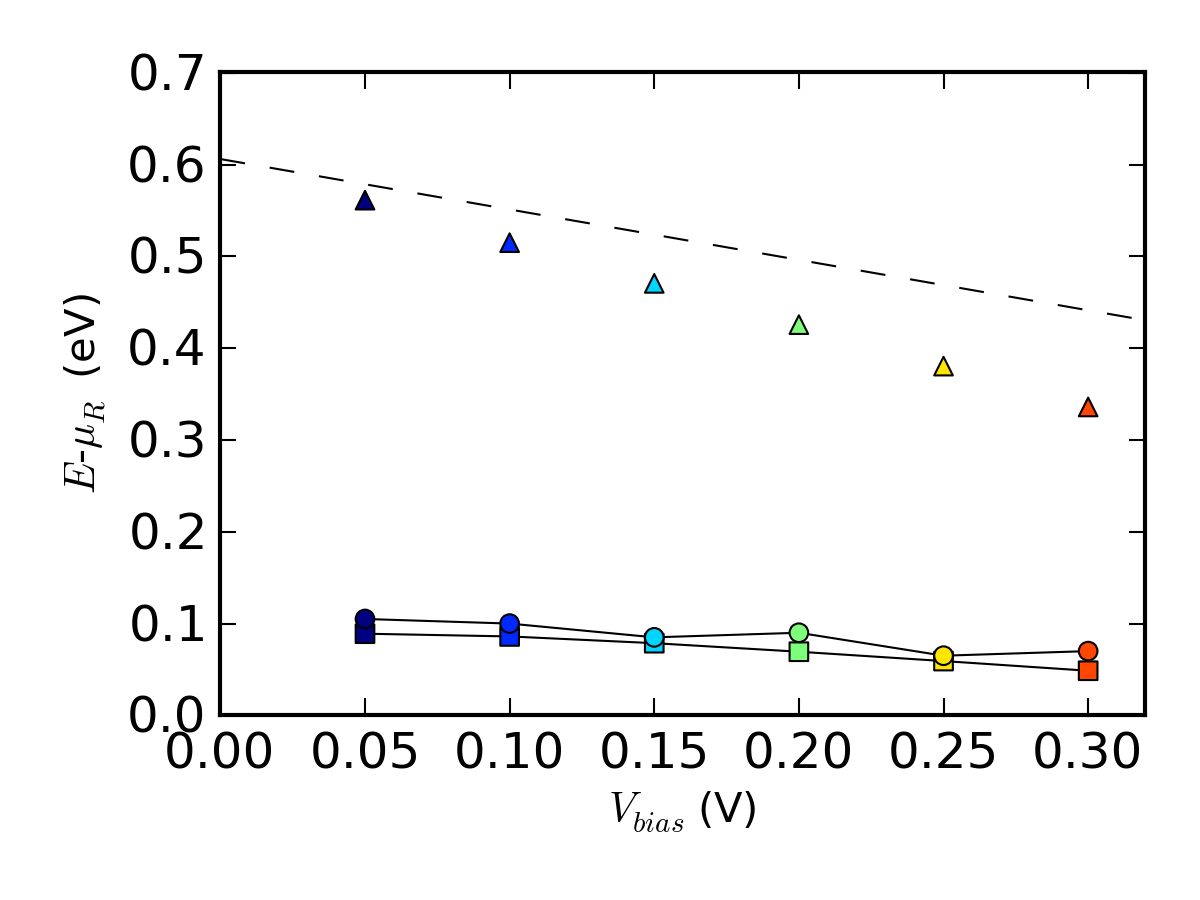
上图显示,每个偏压点处的估算 AE 肖特基势垒(正方形)值都要低于热离子发射势垒(三角形)。这是因为AE法忽略了隧道效应的贡献,因此隐含假设最大谱电流(圆圈)出现在势垒高度能量处。另外,我们可以看出热离子发射势垒随分析偏压变化的曲线(虚线)与模拟的势垒高度的变化趋势(三角形)吻合良好。
电流差别的说明
模拟计算得到的电流要比当前报道在已出版文献 [cSMB+16] 中的电流小两个数量级。这是因为使用了两个有略微差别的 Ag 电极:在界面处两个表面拼接后,文章中所用的银电极没有弛豫,因此与本案例相比,文章中的电极相对被压缩了。通过比较电极的能带结构可以看出,弛豫对电极的电子结构产生了影响。
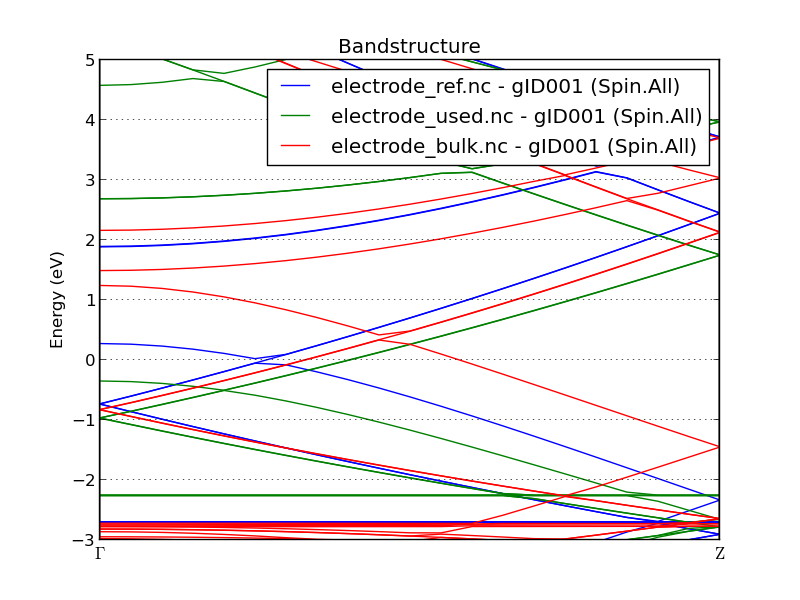
在上图中,“electrode_ref.nc” 是指文章中使用的电极,“eletrode_used.nc” 表示本例用到的电极,而 “electrode_bulk.nc” 则是完全未发生形变的银电极。比较前两个,您会发现一般拉伸电极会导致其具有更低的能量水平。本例研究中用到的电极在偏压窗口具有较少的 k 点(和态)可用,因此产生了较小的输运和较低的电流。然而,如果比较前两个和未形变银的能带结构,我们可以看到在费米能级附近的主要特点是保留着这里用到的两个电极和出版物中用到的一个。
参考
- [cBBR88] | A. Baldereschi, S. Baroni, and R. Resta. Band offsets in lattice-matched heterojunctions: A model and first-principles calculations for GaAs/AlAs. Phys. Rev. Lett., 61:734–737, Aug 1988.
doi:10.1103/PhysRevLett.61.734. - [cBML13] | R. Balsano, A. Matsubayashi, and V. P. LaBella. Schottky barrier height measurements of cu/si(001), ag/si(001), and au/si(001) interfaces utilizing ballistic electron emission microscopy and ballistic hole emission microscopy. AIP Advances, 3:112110, 2013. doi:10.1063/1.4831756.
- [cGASL10] | J. J. Garramone, J. R. Abel, I. L. Sitnitsky, and V. P. LaBella. Hot-electron transport studies of the Ag/Si(001) interface using ballistic electron emission microscopy. Journal of Vacuum Science & Technology A, 28:643, 2010. doi:10.1116/1.3397795.
- [cKP05] | E. Kasper and D. J. Paul. Silicon Quantum Integrated Circuits. Springer-Verlag Berlin Heidelberg, 2005. doi:10.1007/b137494.
- [cKit04] | C. Kittel. Introduction to Solid State Physics. Wiley, 8th edition, 2004.
- [cSMB+16] | (1, 2, 3, 4) D. Stradi, U. Martinez, A. Blom, M. Brandbyge, and K. Stokbro. General atomistic approach for modeling metal-semiconductor interfaces using density functional theory and nonequilibrium green’s function. Phys. Rev. B, 93:155302, Apr 2016.doi:10.1103/PhysRevB.93.155302.
- [cSK06] | (1, 2) S. M. Sze and K. N. Kwok. Physics of Semiconductor Devices. Wiley, 3rd edition, 2006.
- [cTB09] | F. Tran and P. Blaha. Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett., 102:226401, 2009. doi:10.1103/PhysRevLett.102.226401.