
概述
QuantumATK 材料与器件模拟平台包含的 DFT-PlaneWave 全功能平面波密度泛函理论计算引擎与平台的图形用户界面 NanoLab 完美集成,可能是目前最灵活和友好的平面波程序。DFT-PlaneWave 实现了赝势和平面波基组相结合的第一性原理密度泛函理论电子结构计算方法。DFT-PlaneWave 使用内置的模守恒赝势,涵盖了元素周期表中绝大部分的元素。ATK-DFT 实现了众多版本的局域密度近似(LDA)和广义梯度近似(GGA)的交换关联函数。meta-GGA、DFT-1/2、HSE06杂化泛函等则可以准确、快速地计算半导体、绝缘体材料的带隙。
更多功能介绍详见 QuantumATK功能列表。
- 基于平面波基组的 DFT 计算引擎
- 完全自主开发的代码,完美集成于图形界面环境
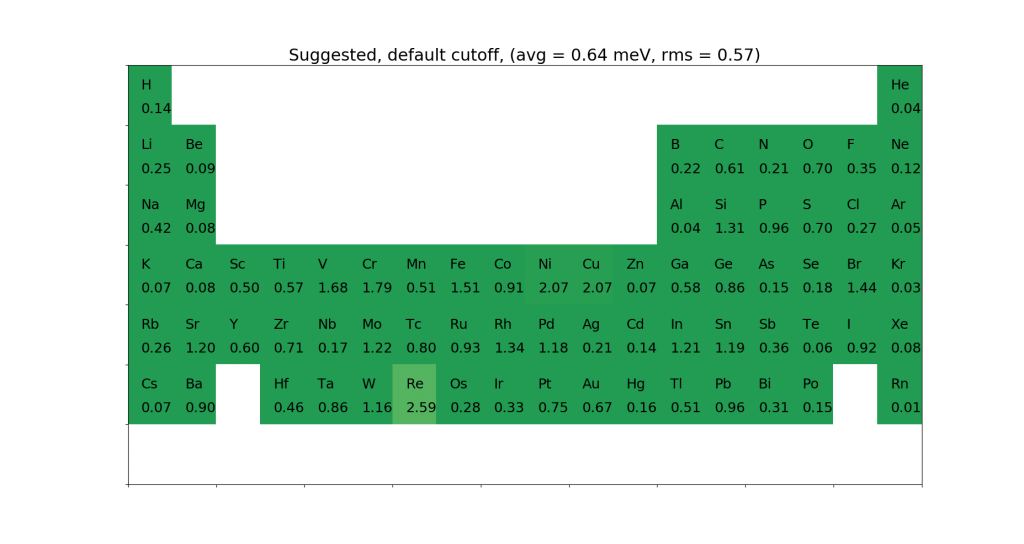
- 针对所有元素都有默认的网格截断能设置
- 支持杂化泛函(使用新颖的 Adaptively Compressed Exchange(ACE)算符方法,比传统方计算性能更佳)
- 可选模守恒 Troullier-Martins 赝势
- FHI/SG15/PseudoDojo势,用于周期表几乎全部元素,很多元素支持半芯势
- PseudoDojo 和 SG15 势是全相对论的
- 可选Projector Augmented Wave(PAW)势
- 使用更小的平面波截断能计算
- GPAW/JTH两套PAW势数据,支持稀土元素计算
- 超过 300 种 LDA/GGA/metaGGA 交换相关泛函(libXC)
- LDA:HL,PW,PZ,RPA,WIGNER,XA 等
- GGA:BLYP,BP86,BPW91,PBE,PBES,PW91,RPBE,XLYP 等
- metaGGA:SCAN
- 多种杂化泛函HSE06、B3LYP、B3LYP5等供选择
- 用于半导体和绝缘体精确带隙计算的方法
- MetaGGA
- DFT+1/2
- 经验的“赝势投影算符移动”(Pseudopotential Projector Shift,PPS)方法(内含 Si 和 Ge 的参数)
- 范德华力模型(DFT-D2 和 DFT-D3)
- 非共线、限制性和非限制性的自旋极化计算
- 自旋-轨道耦合
- 本征值求解
- 解析计算力和张力
DFT-PlaneWave的计算性能
计算精度
无论是使用 PseudoDojo 赝势还是PAW势,DFT-PlaneWave 均可以得到更高精度计算结果。
计算速度与并行化
DFT-PlaneWave适合小体系的高精度计算,支持多级别并行化:
- NEB 路径中间结构等多计算并行
- k 点并行
- 平面波态并行(多进程处理一个 k 点)
- 能带和态密度计算
- 自动确定每 k 点的最佳并行进程数
DFT-PlaneWave支持的计算类型
- 用于多种模型计算
- 分子(Molecule)
- 块体材料(Bulk)
- 计算各种材料性质
- 电子态
- 能带、态密度
- 投影能带和投影态密度(投影在自旋(上/下),自旋(x/y/z),原子位,原子组标签、元素、壳层、轨道等)
- 有效能带
- 本征值
- 能量与力
- 力、张力优化
- 动力学与动态性质
- 分子动力学
- NEB过渡态与反应路径搜索
- 声子与热性质
- 输运性质
- 有效质量
- LocalBandStructure
- 光学与谱学
- 光谱与介电函数谱
- 核磁共振谱与电场梯度
- 电子态
与其他平面波代码相比的优势
- 用户友好
- 与 NanoLab 图形用户界面和 Python 接口完全集成,方便设置复杂计算和编程
- 自洽、结构优化、能带、动力学矩阵等计算都可以在一个计算流程中完成(单个脚本)
- 不需要在并行读写数据时使用相同的进程数
- 使用 ACE 的快速 HSE 方法
- 目前 VASP 里面没有
- HSE 能带计算(QE 里需要在自洽里包含全部 k 点)
- 默认从波函数重启计算,效率高(可以关闭以节约内存)
- 使用其他方法重启、初始化计算可以提高性能
- 使用 LCAO DFT 计算作为初始电子密度(大体系、非周期体系有用)
- 使用不同的 cutoff 重启计算,测试收敛性时有用
- 非共线计算可以从共线或非共线计算得到的电子密度出发,旋转成非共线形式。磁各项异性计算有用
- 非自洽计算:密度-有效势-哈密顿量-本征值/波函数
- 第一个实现了PPCG算法的代码
- 使用Kerker预处理工具改进使用平面波DFT对片层(Slab)结构计算的收敛性
- 支持 Poisson solver 的非周期边界条件,用于slab,金属区域,电场等等
- 赝势默认 cutoff 值,Delta test 结果非常好
- 可以用于 Synopsys 的 Sentaurus Materials Workbench 的计算